
Mobile Genetic Elements Drive the Antibiotic Resistome Alteration in Freshwater Shrimp Aquaculture

1
Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Nanjing 210044, China
2
State Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023, China
3
Nanjing Jiangdao Institute of Environmental Research Co., Ltd., Nanjing 210019, China
*
Author to whom correspondence should be addressed.
Water 2021, 13(11), 1461; https://doi.org/10.3390/w13111461
Submission received: 20 April 2021
/
Revised: 13 May 2021
/
Accepted: 19 May 2021
/
Published: 23 May 2021
(This article belongs to the Section Biodiversity and Functionality of Aquatic Ecosystems)
Abstract
:Shrimp aquaculture environments are a natural reservoir of multiple antibiotic resistance genes (ARGs) due to the overuse of antibiotics. Nowadays, the prevalence of these kinds of emerging contaminants in shrimp aquaculture environments is still unclear. In this study, high-throughput sequencing techniques were used to analyze the distribution of ARGs and mobile genetic elements (MGEs), bacterial communities, and their correlations in water and sediment samples in two types of typical shrimp (Procambarus clarkii and Macrobrachium rosenbergii) freshwater aquaculture environments. A total of 318 ARG subtypes within 19 ARG types were detected in all the samples. The biodiversity and relative abundance of ARGs in sediment samples showed much higher levels compared to water samples from all ponds in the study area. Bacitracin (17.44–82.82%) and multidrug (8.57–49.70%) were dominant ARG types in P. clarkii ponds, while sulfonamide (26.33–39.59%) and bacitracin (12.75–37.11%) were dominant ARG types in M. rosenbergii ponds. Network analysis underlined the complex co-occurrence patterns between bacterial communities and ARGs. Proteobacteria, Cyanobacteria, and Actinobacteria exhibited a high abundance in all samples, in which C39 (OTU25355) and Hydrogenophaga (OTU162961) played important roles in the dissemination of and variation in ARGs based on their strong connections between ARGs and bacterial communities. Furthermore, pathogens (e.g., Aeromonadaceae (OTU195200) and Microbacteriaceae (OTU16033)), which were potential hosts for various ARGs, may accelerate the propagation of ARGs and be harmful to human health via horizontal gene transfer mediated by MGEs. Variation partitioning analysis further confirmed that MGEs were the most crucial contributor (74.76%) driving the resistome alteration. This study may help us to understand the non-ignorable correlations among ARGs, bacterial diversity, and MGEs in the shrimp freshwater aquaculture environments.
1. Introduction
The increasing prevalence of antibiotic resistance genes (ARGs) caused by the use of antibiotics has been recognized as serious emerging contaminants [1]. These scenarios provide ideal conditions for the emergence and selection of antibiotic resistant bacteria (ARB) in environmental media and could easily stimulate horizontal gene transfer (HGT) [2]. Nowadays, ARGs widely exist in most aquatic environments [3], animal feedlots [4], drinking water [5], wastewater treatment plants [6], and even air [7]. The uncontrolled use of antimicrobial agents drives the development and dissemination of ARGs among different microorganisms, in which the increasing levels of human and animal pathogens obviously pose serious risks to the environment [8,9]. The total global aquaculture production exceeded global fishery production by over 18.32 million tonnes during 2017 [10]. The vigorous development of the aquaculture industry was inevitably accompanied by the massive use of antibiotics worldwide to maximize the speed of growth and intensity of production [11], although the use of some antibiotics was banned [12]. Therefore, shrimp aquaculture environments are a reservoir of ARGs [11,13]. China, the largest producer and exporter of aquatic products, has released a large number of antibiotics belonging to different categories in the freshwater and saltwater aquaculture environments, leading to an increasing number of ARB and virulent pathogens, and to ARGs-related pollution [14,15]. The formulation of the “One Health” concept has further emphasized the inevitable relationships between human health, animals (e.g., aquaculture, livestock, and wildlife), and the environment, driving people to explore the associations between ARGs and microorganisms in the aquaculture environment [16]. Polymerase chain reaction (PCR)-based approaches and high-throughput sequencing (HTS)-based approaches have been widely applied to determine and quantify ARGs in aquaculture environments in previous studies. Based on known DNA sequences, a certain number of different types of ARGs from environmental samples could be easily obtained using PCR-based approaches. For instance, 13 types of ARGs, including tetracycline resistance genes, sulfonamide resistance genes, and plasmid-mediated quinolone resistance genes in fish ponds were analyzed using PCR and quantitative PCR (qPCR) methods [17]. Moreover, the PCR technique could be combined with antibiotic susceptibility testing to explore ARG patterns in aquaculture environments [18]. A recent study demonstrated that culturable bacteria in the shrimp hepatopancreas and culture ponds were able to carry multiple ARGs, including sul1, sul2, floR, strA, and gyrA, which varied with the phase of the aquatic culture [19]. In addition to natural aquaculture environments, great amounts of Vibrio parahaemolyticus strains isolated from commonly consumed fresh shrimps (Litopenaeus vannamei, Macrobrachium rosenbergii, Penaeus monodon, and Exopalaemon carinicauda) in fish markets were also observed to display multidrug resistance [20]. This combined method cannot indicate the ARGs level of the whole bacterial community, which is restricted by the cultivability of different bacteria. Similarly, the improved high-throughput qPCR can more effectively analyze the ARG distribution in both biological and environmental samples [21]. However, the use of such a traditional method limited these studies only to investigating target ARGs with known primers for amplification; thus, they could not perform a comprehensive analysis of all ARGs in aquaculture environments [16].
Alternatively, HTS-based approaches have been applied to profile the abundance and diversity of bacterial communities and functional genes in recent years, therefore bringing great convenience to the exploration of ARB or ARGs in biotic/abiotic samples. Thereinto, 16S rRNA gene high-throughput sequencing was usually used to analyze the structure and diversity of bacterial communities. It is often combined with q-PCR in previous studies [22]. For instance, Zeng et al. (2019) [23] revealed the relationship between antibiotic feeding and the abundance of ARGs, as well as the microbiota shift in the shrimp intestine. However, there are few studies on the simultaneous application of HTS-based approaches to the analysis of both the bacterial community and ARGs in shrimp aquaculture environments. The diversity and abundance of ARGs in shrimp gut samples, the water, and sediment samples of the shrimp aquaculture environment were comprehensively investigated using HTS-based metagenomic technology [24]. Plasmid metagenomic sequencing analysis has also been applied in the intestinal bacteria of Penaeus vannamei, but the plasmid DNA samples used for sequencing were extracted from selected ARB rather than all bacteria in samples [25]. However, none of these studies have taken into account the differences between shrimp species. Apart from that, the distribution of ARGs and their associations with bacterial communities/diversity and MGEs in different shrimp aquaculture environments remain unknown.
In this study, the ARG and MGE distribution, diversity, and structure of bacterial communities and their associations were comprehensively investigated via HTS-based metagenomic approaches focusing on two types of typical shrimp (Procambarus clarkii and Macrobrachium rosenbergii) freshwater aquaculture environments. Our work aims to reveal the ARG distribution in water and sediment samples and their associations with bacteria and MGEs. The findings will help to provide comprehensive and reliable information to understand the prevalence of ARGs in freshwater aquaculture systems better.
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
Four P. clarkii aquaculture ponds and three M. rosenbergii aquaculture ponds in Jiangsu Province, China, were selected for water and sediment sampling. Detailed information about the sampling sites is summarized in Table 1. Water and sediment samples from two corresponding downstream rivers in Xuyi and Gaoyou were also collected synchronously. Water samples were collected using sterile bottles from sites that were 0.5 m under the water surface at the cross corners in each pond. Sediment samples were collected from sites that were 0–1 cm below the sludge surface using a grab sampler at the same cross corner of each pond. All the samples were stored in the dark in a portable ice box (50 L) and transferred to the laboratory immediately. Water samples (150 mL for each) filtered by a sterile 0.22 μm membrane filter (GTTP04700, Merck Millipore, Burlington, MA, USA) and sediment samples (0.25 g wet mass for each) were stored at −80 °C before DNA extraction. For each site, three reduplicates were prepared for DNA extraction and high-throughput sequencing. The PowerWater DNA Isolation Kit and the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA) were used for total genomic DNA extraction from the water and sediment samples, respectively. All the sample IDs were designated by species name (in italic), city abbreviation, downstream river (if necessary), sampling site number in the same city, and water/sediment type. For example, PXYR1w refers to the DNA sample of water sampled from the downstream river of the first site of P. clarkii aquaculture ponds in Xuyi.
2.2. High-Throughput Sequencing
To analyze the diversity and abundance of the ARGs and MGEs, DNA samples from each sampling site were sent to the Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) for library construction and Illumina shotgun high-throughput sequencing using the PE 150 (paired-end sequencing, 150 bp) sequencing strategy.
The 16S rRNA gene sequencing was applied to explore the diversity and structure of bacterial communities in aquaculture environments. The hypervariable region (V3–V4) of the 16S rRNA gene was amplified with a set of universal forward (5′-CCTACGGGNGGCWGCAG-3′) and reverse (5′-GACTACHVGGGTATCTAATCC-3′) primers. The following PCR program was performed: initial denaturation at 98 °C for 5 min, followed by 25 cycles at 98 °C for 30 s, 50 °C for 30 s, 72 °C for 40 s, and a final elongation at 72 °C for 10 min. The purified amplicon products were pooled together with an equal amount of each sample and subsequently sent to Sangon Biotech (Shanghai) Co., Ltd. for bacterial 16S rRNA gene sequencing on the Illumina Miseq platform (Illumina, San Diego, CA, USA).
2.3. Bioinformatics Analysis
Raw reads contaminated by the adapter or those containing three or more ambiguous nucleotides (“N”) with an average quality score below 20 or with a length less than 50 bp were removed using Trimmomatic [26]. Clean metagenomic sequencing reads were searched for the presence of ARGs using ARGs-OAP 2.0 [27] with default parameters. MGEs (integrons, insertion sequences (ISs), and plasmids) were quantified using metagenomic methods according to our previous study [28]. To identify the potential pathogens, MetaPhlAn2 was used to conduct taxonomic classification at the species level (or at strain level) [29], and the results of the MetaPhlAn2 were compared to the bacterial pathogen database which contains 538 pathogenic species [30].
The generated sequencing reads were executed for quality control using the Mothur software [31]. The high-quality reads were clustered into Operational Taxonomic Units (OTUs) and α-diversity indices (Chao1, Shannon, and Simpson) with a threshold of 97% using QIIME [32] to estimate the community richness. The taxonomic assignment of the representative sequences was performed using a default method with the RDP Classifier at an 80% confidence threshold [33].
2.4. Statistical Analysis
The relative abundance of ARGs (%) was calculated as the proportion of the annotated reads of one ARG out of the total reads of all ARGs. The relative abundance of MGEs investigated by aligning quality-filtered reads against the MGEs databases was determined using the unit of ‘ppm’—namely, one hit in 1,000,000 aligned sequencing reads.
In order to investigate the co-occurrence patterns between bacteria and ARGs, the network analysis based on Spearman’s correlation coefficient (R) was performed using Gephi as described in the previous study [34,35,36]. Connections between two bacterial genera, two ARG subtypes, or bacterial genera and an ARG subtype with a significant positive correlation (R ≥ 0.60, p ≤ 0.01) were selected to conduct the network visualization. Variation partitioning analysis (VPA) was further conducted to determine the contribution of bacterial biodiversity and MGEs to the variations in ARGs using R software and the ‘vegan’ package following the method described by Jia et al. (2015) [5].
3. Results
3.1. Abundance and Diversity of ARGs in the Shrimp Aquaculture Environment
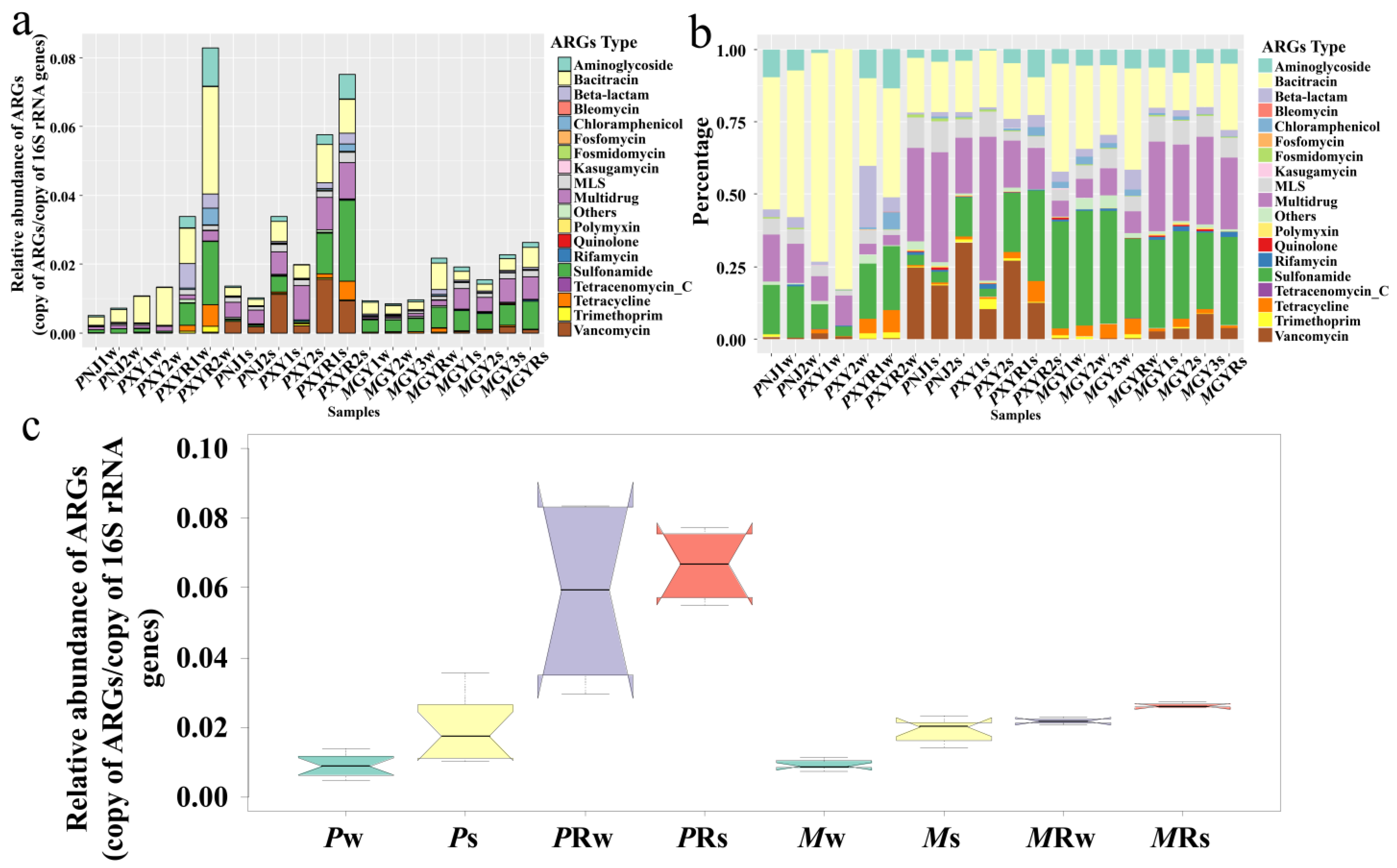
As shown in Figure 1, a total of 318 ARG subtypes within 19 ARG types were identified in all samples. In general, the relative abundance of ARGs in sediment samples was significantly higher than that in the corresponding water samples (Figure 1a,c), indicating that pond sediment is a major reservoir of ARGs. Bacitracin and sulfonamides were predominant in all water samples, while multidrug and vancomycin were predominant in all sediment samples (Figure 1b). Meanwhile, the relative abundance of ARGs in the downstream section of the river was higher than that in shrimp ponds, consistent with the tendency shown in Figure 1c. In P. clarkii aquaculture ponds, multidrug was predominant in sediment samples, with percentages ranging from 19.34% to 49.70%, followed by vancomycin (10.39–33.37%), bacitracin (17.44–19.49%), and macrolide-lincosamide-streptogramin (M-L-S) (6.20–10.61%). In M. rosenbergii aquaculture ponds, sulfonamides (36.92–39.59%) and bacitracin (23.99–37.11%) were dominant in water samples, while multidrug (26.44–30.99%), sulfonamides (26.33–30.45%), and bacitracin (12.75–15.09%) were the main ARG types in sediment samples.
Among all the genes detected, bacA (1.40 × 10−3–3.21 × 10−2 copy of ARGs/copy of 16S rRNA genes), macB (1.90 × 10−5–1.94 × 10−3 copy of ARGs/copy of 16S rRNA genes), transporter (1.08×10−5–5.72×10−3 copy of ARGs/copy of 16S rRNA genes), and sul1 (9.05 × 10−5–1.93 × 10−2 copy of ARGs/copy of 16S rRNA genes) were found in all samples (Figure S1). Bacitracin (bacA), multidrug (transporter), sulfonamide (sul1 and sul2), and vancomycin (vanR) were the main four ARG subtypes in shrimp aquaculture environments. AadA, OXA-21, floR, macB, sul1, tetC, and dfrA1 were relatively the most abundant subtypes in aminoglycoside, beta-lactam, chloramphenicol, M-L-S, sulfonamide, tetracycline, and trimethoprim ARGs, respectively.
3.2. Diversity and Structure of Bacterial Communities in the Shrimp Aquaculture Environment
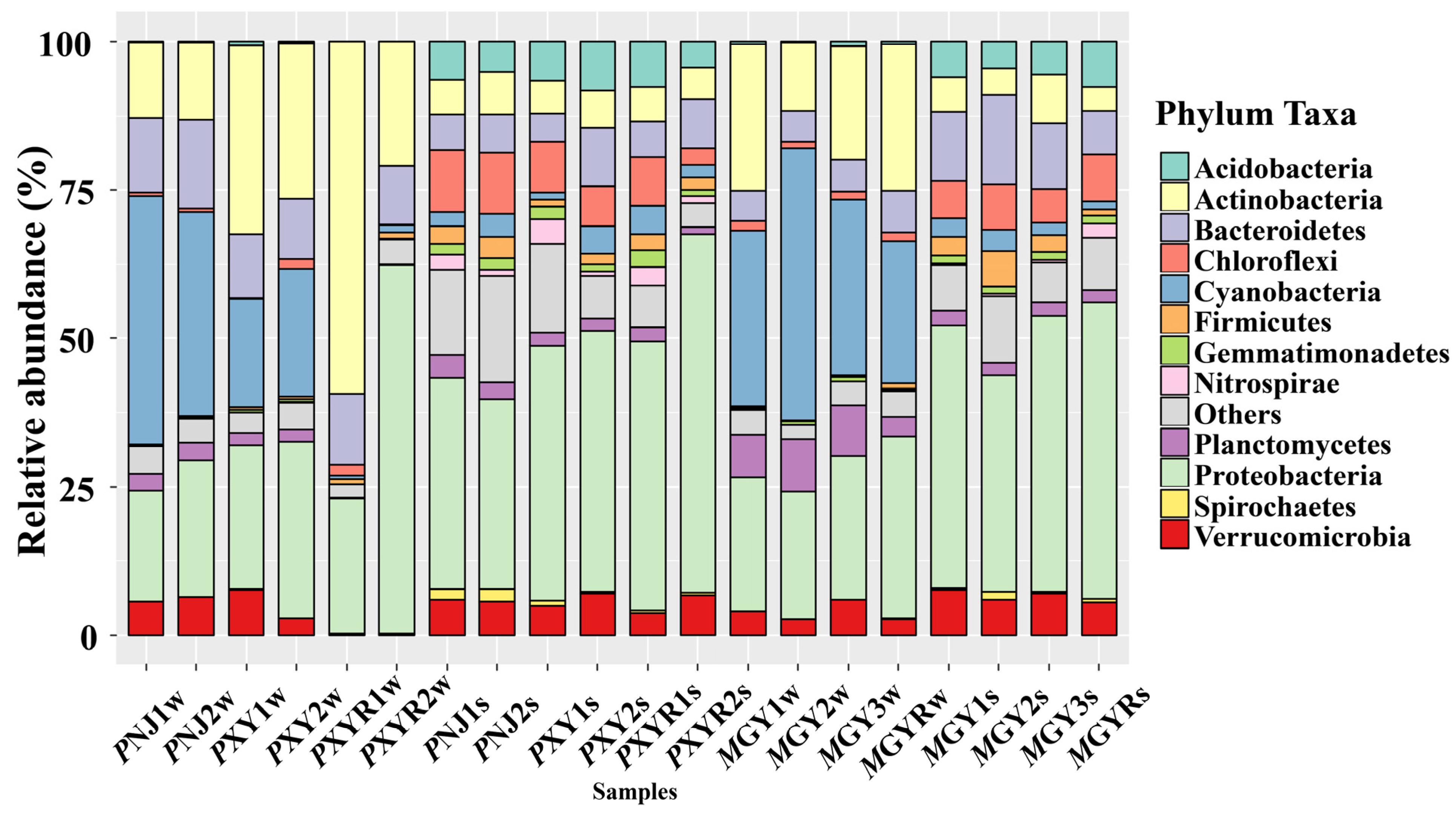
According to Figure 2, the bacterial community structures present differences at the phylum level. Proteobacteria were ubiquitous in all environmental samples, and they were relatively more abundant in sediment samples than those in water samples. However, the relative abundance of Cyanobacteria and Actinobacteria in all ponds was higher than in the corresponding sediment samples. The dominant phylum in the water samples of P. clarkia ponds was Cyanobacteria, while Proteobacteria and Chloroflexi were most abundant in sediment samples. On the other hand, Cyanobacteria was the most dominant phylum in water samples in M. rosenbergii ponds, followed by Proteobacteria and Actinobacteria. Proteobacteria and Bacteroidetes were the most abundant in sediment samples in M. rosenbergii ponds.
The number of reads, OTUs, Chao1 richness estimators, and the Shannon diversity index are listed in Table 2. The OTUs in sediment samples were significantly higher than those in water samples in all shrimp ponds (p < 0.01), except for XY1. The heatmap (Figure S2) also obviously shows that the samples of water and sediment from different regions clustered separately, indicating that the bacterial communities in sediments had a higher richness and diversity than those in water in seven shrimp ponds and the downstream sections of rivers in two cities. In terms of P. clarkia ponds, the most abundant OTUs in sediment samples were Sinobacteraceae (OTU77241), Alcaligenaceae (OTU200907), and Stramenopiles (OTU140157) (Figure S2). As for M. rosenbergii ponds, Microcystis (OTU52886) and Mycobacterium (OTU42294) were the most abundant OTUs in water samples, whereas the dominant OTUs in sediment samples were Dechloromonas (OTU228778) and Sinobacteraceae (OTU77241). Meanwhile, the abundance of C39 (OTU25355) in downstream rivers (0.0031–22.5362 ppm) was significantly higher than that in the corresponding ponds (0.0001–0.0196 ppm), regardless of the environmental media.
3.3. Linkages between Bacterial Community (Including Pathogens) and ARGs
The correlations and co-occurrence patterns between microbial taxa and ARG subtypes were further revealed by the network analysis shown in Figure 3a, which could be separated into 38 OTUs and ten antibiotic classes according to the ARGs: aminoglycoside (aadA, aac(6′)-I, aph(3′′)-I, aph(6′)-I), bacitracin (bacA), beta-lactam (class A beta-lactamase, class B beta-lactamase, OXA-21, VEB-1), chloramphenicol (catB, chloramphenicol exporter, floR), M-L-S (ereA, ermF, macB), multidrug (acrB, mexB, mexF, ABC_transporter, transporter, ompR), sulfonamide (sul1, sul2), tetracycline (tet44, tetA, tetC, tetG, tetM, tetX), trimethoprim (dfrA1, dfrA17), and vancomycin (vanR). The network suggested that C39 (OTU25355) was the most active bacterium, potentially carrying 12 ARG subtypes simultaneously, including aadA, tetX, tetG, tet44, sul1, sul2, floR, aph(6′)-I, class A beta-lactamase, OXA-21, VEB-1, and chloramphenicol exporter. Hydrogenophaga (OTU162961) was significantly positively related to six ARG subtypes and three OTUs (R > 0.60, p < 0.01), and therefore was possibly an active intermediate host in the transfer of ARGs. Moreover, Dechloromonas (OTU228778), Sinobacteraceae (OTU77241), Alcaligenaceae (OTU200907), and Microbacteriaceae (OTU16033) also played important roles in the correlation between bacterial communities and ARGs as potential hosts for a variety of ARG subtypes that maintain relationships with other important OTUs as well. On the other hand, the ARGs of beta-lactam (C39 (OTU25355), Hydrogenophaga (OTU162961), Microbacteriaceae (OTU118139 and OTU16033)), chloramphenicol (C39 (OTU25355), Hydrogenophaga (OTU162961), Microbacteriaceae (OTU16033)), M-L-S (Alcaligenaceae (OTU200907), Dechloromonas (OTU228778), Sinobacteraceae (OTU77241)), and multidrug (Alcaligenaceae (OTU200907), Dechloromonas (OTU228778), Sinobacteraceae (OTU77241)) had more than three common potential host bacteria, indicating that those ARGs may be transferred to diverse bacterial hosts more easily through HGT. Therefore, the strong correlations among microbial taxa shown in Figure 3a would be considered to accelerate the transfer of ARGs.
Furthermore, the diversity and relative abundance of potential pathogens in sediment samples of shrimp ponds were higher than those in water samples (Figure 3b), consistent with the trends of ARGs and bacterial communities. Aeromonas veronii was detected with the highest relative abundance (1.880–174.023‰) in two shrimp ponds. The relative abundance of pathogens in M. rosenbergii ponds was generally slightly higher than that in P. clarkii ponds, while Enterobacter cloacae, Clostridium bifermentans, and Bacillus pumilus were also abundant in the sediment samples of P. clarkii ponds. Regarding M. rosenbergii ponds, A. veronii (2.160–174.023‰), Vibrio cholerae (0.730–64.980‰), and E. cloacae (0.090–17.073‰) were the major pathogens. Furthermore, it is worth noting that Aeromonas spp., including A. veroni, Aeromonas caviae, and Aeromonas hydrophila, mainly affiliated with the Aeromonadaceae (OTU195200), were positively related to Hydrogenophaga (OTU162961) and Microbacteriaceae (OTU16033), which were the main OTUs and potential hosts of various ARGs. In particular, Microbacteriaceae (OTU16033) may have had the most vital contribution in carrying ARGs into the bacterial community and assisting in spreading, as they are an intermediate host that are both potential hosts for various ARGs and significantly positively correlated with multiple bacteria.
3.4. Correlations among Biodiversity, MGEs, and ARGs
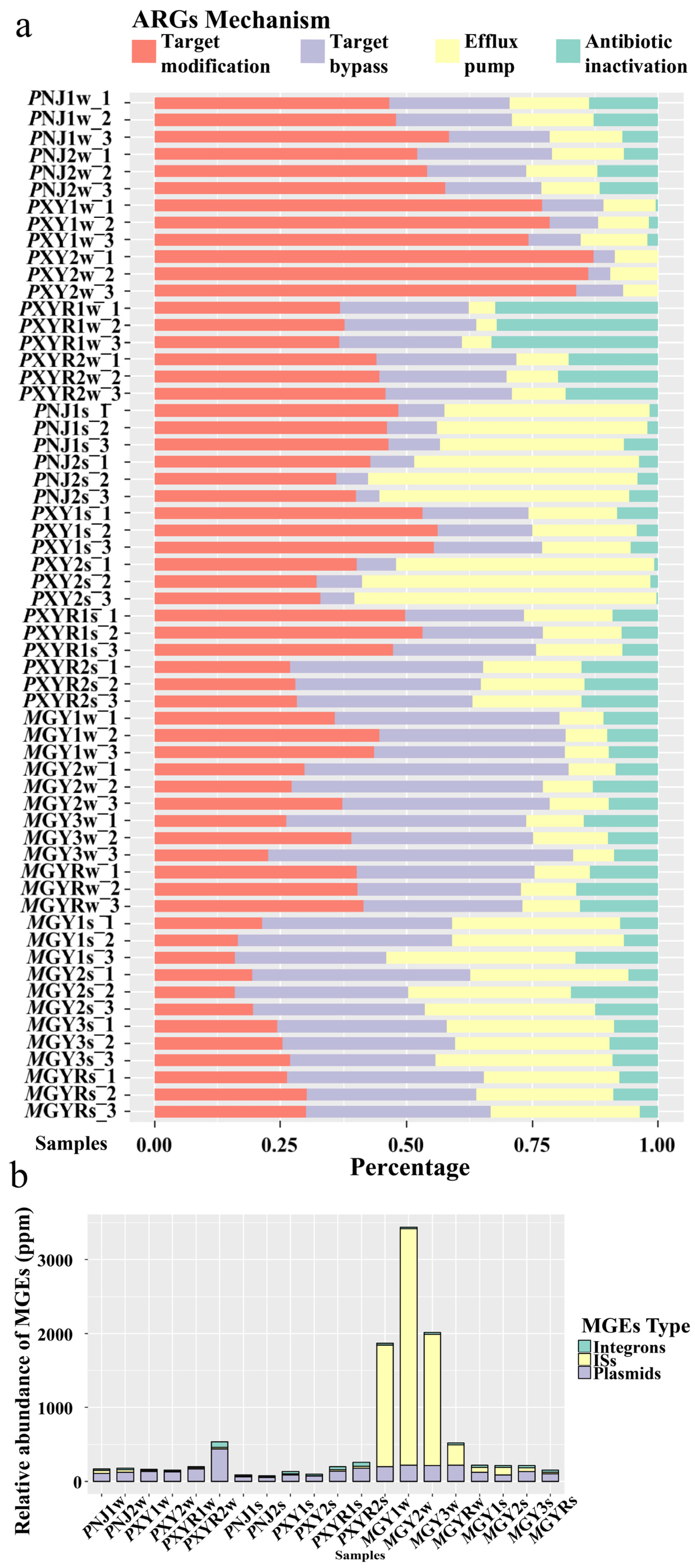
The relative abundance of different resistance mechanisms of ARGs in all environmental samples is shown in Figure 4a. The most predominant resistance mechanism varied by shrimp type. Target modification (32.2–87.2%) was the predominant resistance mechanism in the P. clarkii aquaculture environment, while target bypass (28.9–60.5%) was the major resistance mechanism in the M. rosenbergii aquaculture environment. An efflux pump accounted for a higher proportion in sediment samples (30.7–37.7%) than that in water samples (8.1–15.0%) in both types of shrimp ponds.
Figure 4b shows the relative abundance of integrons, insert sequences (ISs), and plasmids in all water and sediment samples. Overall, three types of MGEs were detected in all samples, and the abundance of MGEs in water samples in M. rosenbergii ponds (1870.8–3439.9 ppm) was significantly higher than in other samples, while no significant difference was observed in other samples. Plasmids (48.7–135.0 ppm) were the most abundant MGEs in the P. clarkii aquaculture area, while ISs (47.4–3197.9 ppm) were the most prevalent MGEs in M. rosenbergii ponds.
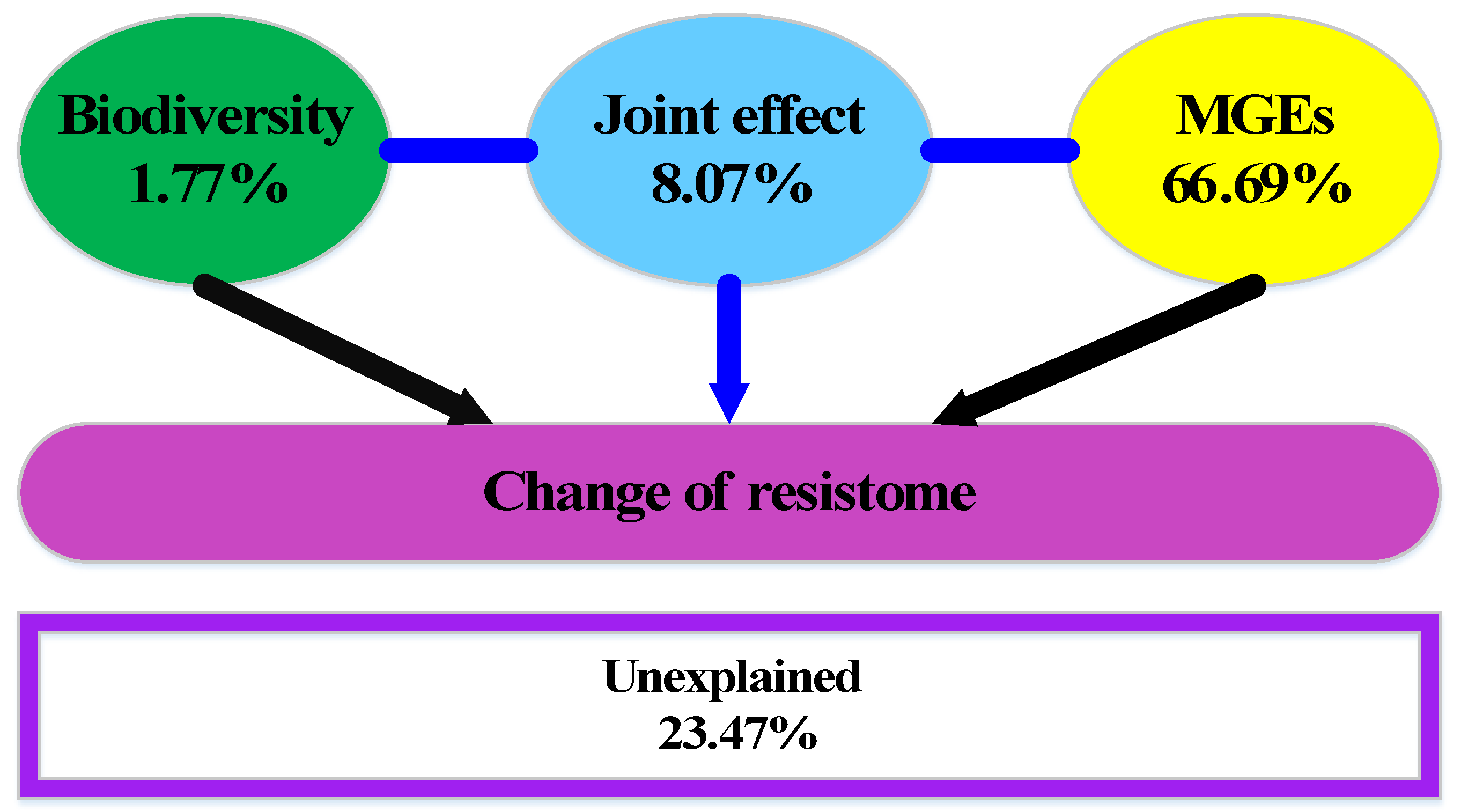
VPA illustrated that 76.53% of the variance in ARGs could be explained by biodiversity and MGEs (integrons, ISs, and plasmids), leaving 23.47% of the variation unexplained (Figure 5). The separate effects of biodiversity and MGEs were obtained by partial redundancy analysis, accounting for 1.77% and 66.69% of the total change in resistome, respectively. MGEs were the most important contributor to the variance of ARGs in the shrimp aquaculture environment, which was much higher than the biodiversity and their joint effects.
4. Discussion
4.1. ARGs Distribution in Freshwater Shrimp Aquaculture Environment
Illumina high-throughput sequencing was used to investigate the abundance and diversity of ARGs in shrimp aquaculture environments. Our results revealed that the relative abundance of ARGs in sediments was significantly higher than that in water samples, which may be related to sediment adsorption, and the input of excrement as a result of the activities of shrimp [37,38]. Bacitracin was the predominant ARG type in all water samples, which may be intrinsic in bacteria [39]. The bactericidal action of bacitracin is a result of interaction with undecaprenyl pyrophosphate [40], which inhibits peptidoglycan synthesis by preventing the lipid carrier from re-entering the reaction cycle of cell wall synthesis [41]. There is evidence that the use of bacitracin was once very prevalent in China’s aquaculture industry as a feed additive [42], even though it has since been banned and listed as an illicit drug. Hence, bacA has been widely detected in aquaculture areas [24], lake water and seawater [43], glaciers [44], and even ready-to-eat food [45]. Here, we suggest that determination of the relative abundance of bacitracin can be used as one of the most important indicators of ARGs contamination in shrimp aquaculture environments. Sul1 and sul2 have been frequently observed in freshwater aquaculture ponds [46,47], marine aquaculture farms [48], and urban parks [49]. In particular, sul1 has been proven to be the predominant ARG in Chinese aquaculture areas, which may be related to the abuse of sulfonamides [42,50]. These abundant ARGs were usually associated with the extensive use of antibiotics in freshwater aquaculture farms in different regions [51]. Hence, the differences between ARGs in this study may be attributed to the differential selection pressures of antibiotics in two types of shrimp aquaculture environments. Furthermore, as the potential host of 12 ARG subtypes, the higher abundance of C39 (OTU25355) was the leading factor that resulted in the higher abundance of ARGs in downstream rivers than in ponds. This tendency has varied in other studies. Typically, the abundance of ARGs in culture ponds is associated with the abundance of ARGs in upstream and downstream environments. For example, the water source is considered as an important medium disseminating ARGs to aquaculture ponds, in which case the concentration of ARGs detected in the downstream river of the ponds may decrease due to water dilution [52]. Another study found that the abundance of ARGs showed no significant differences between upstream, post-treatment, or downstream water samples in freshwater fish farms [53]. These differences could be attributed to differences in the extent of the bacterial community, anthropogenic activities, and the application of antibiotics in different regions [52].
4.2. Associations between ARGs and Bacteria
The diversity and abundance of bacterial communities in sediment samples were much higher than those in water samples, consistent with the tendency of ARGs. Proteobacteria, Cyanobacteria, and Actinobacteria were the most abundant phyla in all samples, and they were also widespread bacteria in urban wastewater and other aquatic environments [54,55]. There is evidence that Proteobacteria can be considered to be a major participant in the spread of ARGs [56], indicating its critical role in the spread of ARGs in aquaculture environments as a result of its relatively high abundance in shrimp aquaculture ponds. Network analysis revealed the existence of complex co-occurrence patterns between bacterial communities and ARG subtypes, suggesting that a non-random pattern also exists in the aquaculture environment [57]. Eight OTUs (Alcaligenaceae (OTU200907), C39 (OTU25355), Dechloromonas (OTU228778), Hydrogenophaga (OTU162961), Microbacteriaceae (OTU118139, OTU14937, and OTU16033) and Sinobacteraceae (OTU77241)) were significantly related to at least one ARG subtype, showing the diversity of potential bacterial hosts in the aquaculture environment. The data show that the potential hosts of ARG subtypes were mainly affiliated with the Proteobacteria and Actinobacteria. For example, Hydrogenophaga (OTU162961) are Gram-negative bacteria, and most Hydrogenophaga species are susceptible to a wide range of antibiotics and bacteriostatic agents [58]. Therefore, Hydrogenophaga play a dominant role in the dissemination of and variation in ARGs in freshwater bodies, regardless of the environmental media, by acting as an important potential intermediate host in the transmission of antibiotic resistance.
Previous studies have suggested that most of pathogenic diseases in aquaculture are often associated with Aeromonas, Vibrio, Streptococcus, Acinetobacter, Pseudomonas, and Clostridium [2,59]. The Aeromonas genus, including A. veronii, A. hydrophila, and A. caviae, can be detected in both marine and freshwater environments as important pathogens in aquaculture [60]. Aeromonas spp. collected from aquaculture farms in the Guangdong province of South China have been detected to carry a class 1 integron comprising several different gene insertion cassettes, suggesting that freshwater animals can serve as a reservoir for pathogenic Aeromonas strains containing multiple drug-resistant integrons [61]. Hence, the Aeromonadaceae (OTU195200), which include both those key pathogenic bacteria and multi-drug resistance genes mentioned in this study, may cause shrimp septicemia and therefore lead to production reduction, and even cause serious hazards to human health by spreading through the food chain [62]. On the other hand, Microbacteriacea (OTU118139 and OTU16033) were found to be significantly positively related to both multiple ARG subtypes and main OTUs. They have been cited as opportunistic human and plant pathogens and may cause skin and soft tissue infections [63,64], showing their non-negligible role in the bacterial community of the shrimp aquaculture environment. In summary, both types of shrimp aquaculture environments contained a higher level of diversity and abundance of potential pathogens compared to crab ponds used in our previous study [28].
4.3. Associations among ARGs, Biodiversity and MGEs
Multiple factors have contributed to the distribution of ARGs. For example, the environmental physicochemical parameters (total organic carbon (TOC) and chemical oxygen demand (COD), etc.) have been verified to have a potential relationship with bacterial communities and ARGs. A strong positive correlation was observed between the levels of sul2 and TOC in shrimp pond water [65]. COD was positively correlated with ARGs in river water as well [66], indicating that altering environmental factors may have a certain impact on the reservoir of ARGs in aquatic environments.
Furthermore, the most prevalent MGEs were different in different shrimp species. Plasmids and ISs were the dominant MGEs in P. clarkii and M. rosenbergii ponds, respectively. Another similar research showed that the relative abundance of MGEs detected in the shrimp gut was between that in the corresponding water and sediment samples [24]. Several studies have shown that plasmids are considered to be the main carrier of ARGs for HGT in the bacterial community as a result of their ability to carry multiple resistance genes [67]. For instance, Aeromonas, Acinetobacter, Citrobacter, and other bacteria were reported to carry plasmids with various sizes [68]. The intI1 gene was also an important indicator affecting the occurrence and horizontal transfer of ARGs in bacteria [69]. Consequently, pathogens that host multiple ARGs would undoubtedly seriously aggravate their hazards to aquatic animals and the environment via accelerating the spread of ARGs and could further unpredictably impact human health.
According to the variation partitioning analysis, MGEs were the main contributions to the variation in ARGs, which was similar to the results reported before, showing that the relative abundance of ARGs was strongly correlated with MGEs (plasmids, integrons, and ISs) [22]. There is further evidence that the abundance of the intI1 gene is significantly correlated with ARGs not only in sediments from aquaculture farms [70] but also in livestock manure from farms [21]. Bacterial communities have been applied to the VPA of ARGs in a large number of studies [57,71,72]. In this study, the biodiversity instead of the bacterial community was used to analyze the contribution of resistome alterations due to its better ability to stand for the whole microbiota. However, our results further emphasized the significant role of MGEs rather than biodiversity in the change in the bacterial resistome in shrimp aquaculture environments.
4.4. Practical Implications of This Study
Our findings revealed the change of resistome in two types of shrimp freshwater aquaculture environments by elaborating the strong and complex associations among ARGs, bacteria, and MGEs. Concentrating on the impacts of more factors to improve the interpretability of ARGs alteration is the main focus of our future work. Moreover, more efforts should be devoted to conduct further studies on ARGs pollution existing in other aquaculture environments. Then, more appropriate and constructive policies could be put forward to reduce the potential risk in freshwater aquaculture environments.
5. Conclusions
In conclusion, we found that the abundance of ARGs, the abundance of MGEs, and the diversity of the bacterial community in sediment samples were all higher than those in the corresponding water samples in P. clarkii and M. rosenbergii aquaculture ponds. Bacitracin, sulfonamides, and multidrug were the three predominant ARGs in the aquaculture environment. The enrichment of C39 (OTU25355) (0.0031–22.5362 ppm) in the bacterial community was the main factor that contributed to the higher relative abundance of ARGs in downstream rivers compared to ponds. The enriched abundance of Proteobacteria (e.g., C39 (OTU25355), Hydrogenophaga (OTU162961), Dechloromonas (OTU228778)) and Actinobacteria (e.g., Microbacteriaceae (OTU118139), Microbacteriaceae (OTU16033)) carrying ARGs resulted in an increase in ARGs. Furthermore, HGT mediated by MGEs (especially plasmids and ISs) was the major driver (74.76%) of ARGs’ alteration. The non-random co-occurrence pattern of ARGs and bacterial communities revealed multiple potential hosts in the aquaculture environment, in which pathogenic bacteria carried a variety of ARGs that could potentially accelerate the spread of ARGs and cause even greater risks to human health.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/w13111461/s1, Figure S1: Relative abundance (copy of ARGs/copy of 16S rRNA genes) of ARG subtypes in all samples, Figure S2: Relative abundance of the major OTUs (>3% in any sample) of all environmental samples collected in P. clarkii and M. rosenbergii aquaculture environments. The color intensity in each panel shows the percentage of each OTU in one sample.
Author Contributions
Conceptualization, H.F. and K.H.; methodology, H.F. and K.H.; formal analysis, K.H.; investigation, N.Y. and J.Y.; writing—original draft preparation, N.Y.; writing—review and editing, H.F., J.Y., K.H., and S.Z.; supervision, K.H.; funding acquisition, K.H., N.Y., and S.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Open Fund of State Key Laboratory of Pollution Control and Resource Reuse, grant number PCRRF20018, the Natural Science Foundation of Jiangsu Province, grant number BK20200816, the Key University Science Research Project of Jiangsu Province, grant number 20KJB610009, and the Science and technology innovation project for overseas students in Nanjing. This work was also sponsored by the Postgraduate Research & Practice Innovation Program of Jiangsu Province, grant number SJCX19_0307.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequences derived from high-throughput sequencing have been deposited in the National Center for Biotechnology Information (NCBI) database. The Illumina shotgun high-throughput sequencing raw data (accession no. PRJNA730203, accessed on 16 May 2021) and 16S rRNA sequencing raw data (accession no. PRJNA729127, accessed on 15 May 2021) are available on the NCBI Sequence Read Archive.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Q.Q.; Yang, G.F.; Sun, K.K.; Tian, G.M.; Jin, R.C. Insights into the effects of bio-augmentation on the granule-based anammox process under continuous oxytetracycline stress: Performance and microflora structure. Chem. Eng. J. 2018, 348, 503–513. [Google Scholar] [CrossRef]
- Santos, L.; Ramos, F. Antimicrobial resistance in aquaculture: Current knowledge and alternatives to tackle the problem. Int. J. Antimicrob. Agents 2018, 52, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Hu, Y.; Cheng, J.; Chen, Y. Research progress on distribution, migration, transformation of antibiotics and antibiotic resistance genes (ARGs) in aquatic environment. Crit. Rev. Biotechnol. 2018, 38, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- He, L.Y.; Liu, Y.S.; Su, H.C.; Zhao, J.L.; Liu, S.S.; Chen, J.; Liu, W.R.; Ying, G.G. Dissemination of antibiotic resistance genes in representative broiler feedlots environments: Identification of indicator ARGs and correlations with environmental variables. Environ. Sci. Technol. 2014, 48, 13120–13129. [Google Scholar] [CrossRef]
- Jia, S.; Shi, P.; Hu, Q.; Li, B.; Zhang, T.; Zhang, X.X. Bacterial community shift drives antibiotic resistance promotion during drinking water chlorination. Environ. Sci. Technol. 2015, 49, 12271–12279. [Google Scholar] [CrossRef]
- Zhang, H.; He, H.; Chen, S.; Huang, T.; Lu, K.; Zhang, Z.; Wang, R.; Zhang, X.; Li, H. Abundance of antibiotic resistance genes and their association with bacterial communities in activated sludge of wastewater treatment plants: Geographical distribution and network analysis. J. Environ. Sci. 2019, 82, 24–38. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Wang, M.; Chen, H.; Yang, Y.; Chen, Q.; Yao, M. Time-resolved spread of antibiotic resistance genes in highly polluted air. Environ. Int. 2019, 127, 333–339. [Google Scholar] [CrossRef]
- Dong, P.; Cui, Q.; Fang, T.; Huang, Y.; Wang, H. Occurrence of antibiotic resistance genes and bacterial pathogens in water and sediment in urban recreational water. J. Environ. Sci. 2019, 77, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Zhao, Y.; Li, B.; Huang, C.L.; Zhang, S.Y.; Yu, S.; Chen, Y.-S.; Zhang, T.; Gillings, M.R.; Su, J.-Q. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Tacon, A.G.J. Trends in global aquaculture and aquafeed production: 2000–2017. Rev. Fish. Sci. 2020, 28, 43–56. [Google Scholar] [CrossRef]
- Watts, J.E.M.; Schreier, H.J.; Lanska, L.; Hale, M.S. The rising tide of antimicrobial resistance in aquaculture: Sources, sinks and solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuah, L.O.; Effarizah, M.E.; Goni, A.M.; Rusul, G. Antibiotic application and emergence of multiple antibiotic resistance (MAR) in global catfish aquaculture. Curr. Environ. Health Rep. 2016, 3, 118–127. [Google Scholar] [CrossRef]
- Zhou, R.; Zeng, S.; Hou, D.; Liu, J.; Weng, S.; He, J.; Huang, Z. Occurrence of human pathogenic bacteria carrying antibiotic resistance genes revealed by metagenomic approach: A case study from an aquatic environment. J. Environ. Sci. 2019, 80, 248–256. [Google Scholar] [CrossRef]
- Liu, X.; Steele, J.C.; Meng, X.Z. Usage, residue, and human health risk of antibiotics in Chinese aquaculture: A review. Environ. Pollut. 2017, 223, 161–169. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Li, Y.; Sun, X.L. Antibiotic resistance of bacteria isolated from shrimp hatcheries and cultural ponds on Donghai Island, China. Mar. Pollut. Bull. 2011, 62, 2299–2307. [Google Scholar] [CrossRef]
- Tiedje, J.M.; Wang, F.; Manaia, C.M.; Virta, M.; Sheng, H.; Ma, L.; Zhang, T.; Topp, E. Antibiotic resistance genes in the human-impacted environment: A One Health perspective. Pedosphere 2019, 29, 273–282. [Google Scholar] [CrossRef]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Li, Y.; Wang, M.; Zeng, Z. Antibiotics, antibiotic resistance genes, and bacterial community composition in fresh water aquaculture environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef]
- Zhou, R.; Zeng, S.; Hou, D.; Liu, J.; Weng, S.; He, J.; Huang, Z. Temporal variation of antibiotic resistance genes carried by culturable bacteria in the shrimp hepatopancreas and shrimp culture pond water. Ecotoxicol. Environ. Saf. 2020, 199, 110738. [Google Scholar] [CrossRef]
- Zhang, R.Q.; Ying, G.G.; Su, H.C.; Zhou, L.J.; Liu, Y.S. Antibiotic resistance and genetic diversity of Escherichia coli isolates from traditional and integrated aquaculture in South China. J. Environ. Sci. Health Part B 2013, 48, 999–1013. [Google Scholar] [CrossRef]
- He, Y.; Jin, L.; Sun, F.; Hu, Q.; Chen, L. Antibiotic and heavy-metal resistance of Vibrio parahaemolyticus isolated from fresh shrimps in Shanghai fish markets, China. Environ. Sci. Pollut. Res. 2016, 23, 15033–15040. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.C.; Lin, Z.J.; Shuai, X.Y.; Zheng, J.; Meng, L.X.; Zhu, L.; Sun, Y.-J.; Shang, W.-C.; Chen, H. Temporal variation and sharing of antibiotic resistance genes between water and wild fish gut in a peri-urban river. J. Environ. Sci. 2021, 103, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Lu, J.; Wu, J.; Zhang, Y.; Zhang, C. Proliferation of antibiotic resistance genes in coastal recirculating mariculture system. Environ. Pollut. 2019, 248, 462–470. [Google Scholar] [CrossRef]
- Zeng, S.; Hou, D.; Liu, J.; Ji, P.; Weng, S.; He, J.; Huang, Z. Antibiotic supplement in feed can perturb the intestinal microbial composition and function in Pacific white shrimp. Appl. Microbiol. Biotechnol. 2019, 103, 3111–3122. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Zhao, Z.; Duan, C.; Chen, H.; Wang, M.; Ren, H.; Yin, Y.; Ye, L. Metagenomic analysis revealed the prevalence of antibiotic resistance genes in the gut and living environment of freshwater shrimp. J. Hazard. Mater. 2018, 350, 10–18. [Google Scholar] [CrossRef]
- Liu, K.; Han, J.; Li, S.; Liu, L.; Lin, W.; Luo, J. Insight into the diversity of antibiotic resistance genes in the intestinal bacteria of shrimp Penaeus vannamei by culture-dependent and independent approaches. Ecotoxicol. Environ. Saf. 2019, 172, 451–459. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Jiang, X.T.; Chai, B.; Li, L.; Yang, Y.; Cole, J.R.; Tjedje, J.M.; Zhang, T. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 2018, 34, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Huang, K.; Yu, J.; Ding, C.; Wang, Z.; Zhao, C.; Yuan, H.; Wang, Z.; Wang, S.; Hu, J.; et al. Metagenomic analysis of bacterial communities and antibiotic resistance genes in the Eriocheir sinensis freshwater aquaculture environment. Chemosphere 2019, 224, 202–211. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.; Gowtage-Sequeria, S.; Evans, B. T16: Quantitative Analysis of the Characteristics of Emerging and Re-Emerging Human Pathogens. Centre for Infectious Diseases, University of Edinburgh. 2015. Available online: http://www.bis.gov.uk/assets/foresight/docs/infectious-diseases/t16.pdf (accessed on 25 July 2015).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Li, B.; Jiang, X.; Yang, Y.; Wells, G.F.; Zhang, T.; Li, X. Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ. Microbiol. 2018, 20, 355–368. [Google Scholar] [CrossRef]
- Ju, F.; Xia, Y.; Guo, F.; Wang, Z.; Zhang, T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ. Microbiol. 2014, 16, 2421–2432. [Google Scholar] [CrossRef]
- Devarajan, N.; Laffite, A.; Graham, N.D.; Meijer, M.; Prabakar, K.; Mubedi, J.I.; Elongo, V.; Mpiana, P.T.; Ibelings, B.W.; Wildi, W.; et al. Accumulation of clinically relevant antibiotic-resistance genes, bacterial load, and metals in freshwater lake sediments in central Europe. Environ. Sci. Technol. 2015, 49, 6528–6537. [Google Scholar] [CrossRef]
- Pruden, A.; Arabi, M.; Storteboom, H.N. Correlation between upstream human activities and riverine antibiotic resistance genes. Environ. Sci. Technol. 2012, 46, 11541–11549. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X.; et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013, 4, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, T.; Margulis, N.G.; Wang, T.; Ye, R.W.; Helmann, J.D. Cell wall stress responses in Bacillus subtilis: The regulatory network of the bacitracin stimulon. Mol. Microbiol. 2003, 50, 1591–1604. [Google Scholar] [CrossRef]
- Siewert, G.; Strominger, J.L. Bacitracin: An inhibitor of the dephosphorylation of lipid pyrophosphate, an intermediate in the biosynthesis of the peptidoglycan of bacterial cell walls. Proc. Natl. Acad. Sci. USA 1967, 57, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Li, Y.; Qi, Z.; Yue, Y.; Min, M.; Peng, S.; Shi, Z.; Gao, Y. Diverse and abundant antibiotic resistance genes from mariculture sites of China’s coastline. Sci. Total Environ. 2018, 630, 117–125. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Song, W.; Du, L.; Ye, C.; Zhao, B.; Liu, W.; Deng, D.; Pan, Y.; Lin, H.; et al. Metagenomic insights into the abundance and composition of resistance genes in aquatic environments: Influence of stratification and geography. Environ. Int. 2019, 127, 371–380. [Google Scholar] [CrossRef]
- Sherpa, M.T.; Najar, I.N.; Das, S.; Thakur, N. Distribution of antibiotic and metal resistance genes in two glaciers of North Sikkim, India. Ecotoxicol. Environ. Saf. 2020, 203, 111037. [Google Scholar] [CrossRef]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 15175. [Google Scholar] [CrossRef]
- Phuong Hoa, P.T.; Nonaka, L.; Hung Viet, P.; Suzuki, S. Detection of the sul1, sul2, and sul3 genes in sulfonamide-resistant bacteria from wastewater and shrimp ponds of north Vietnam. Sci. Total Environ. 2008, 405, 377–384. [Google Scholar] [CrossRef]
- Wang, L.; Su, H.; Hu, X.; Xu, Y.; Xu, W.; Huang, X.; Li, Z.; Cao, Y.; Wen, G. Abundance and removal of antibiotic resistance genes (ARGs) in the rearing environments of intensive shrimp aquaculture in South China. J. Environ. Sci. Health Part B 2019, 54, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ying, C.; Chang, M.J.; Hu, C.H.; Chang, Y.T.; Chao, W.L.; Yeh, S.L.; Chang, S.J.; Hsu, J.T. The effects of marine farm-scale sequentially integrated multi-trophic aquaculture systems on microbial community composition, prevalence of sulfonamide-resistant bacteria and sulfonamide resistance gene sul1. Sci. Total Environ. 2018, 643, 681–691. [Google Scholar] [CrossRef]
- Echeverria-Palencia, C.M.; Thulsiraj, V.; Tran, N.; Ericksen, C.A.; Melendez, I.; Sanchez, M.G.; Walpert, D.; Yuan, T.; Ficara, E.; Senthilkumar, N.; et al. Disparate antibiotic resistance gene quantities revealed across 4 major cities in California: A survey in drinking water, air, and soil at 24 public parks. ACS Omega 2017, 2, 2255–2263. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Ni, M.; Liu, M.; Zheng, Y.; Gu, Z. Occurrence of antibiotics and antibiotic resistance genes in a typical estuary aquaculture region of Hangzhou Bay, China. Mar. Pollut. Bull. 2019, 138, 376–384. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Su, H.; Liu, S.; Hu, X.; Xu, X.; Xu, W.; Xu, Y.; Li, Z.; Wen, G.; Liu, Y.; Cao, Y. Occurrence and temporal variation of antibiotic resistance genes (ARGs) in shrimp aquaculture: ARGs dissemination from farming source to reared organisms. Sci. Total Environ. 2017, 607–608, 357–366. [Google Scholar] [CrossRef]
- Wang, J.H.; Lu, J.; Zhang, Y.X.; Wu, J.; Luo, Y.; Liu, H. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef]
- Becerra-Castro, C.; Macedo, G.; Silva, A.M.T.; Manaia, C.M.; Nunes, O.C. Proteobacteria become predominant during regrowth after water disinfection. Sci. Total Environ. 2016, 573, 313–323. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y.; Wu, J. Continental-scale spatio-temporal distribution of antibiotic resistance genes in coastal waters along coastline of China. Chemosphere 2020, 247, 125908. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic resistance mechanisms in bacteria: Relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef]
- Han, Y.; Wang, J.; Zhao, Z.; Chen, J.; Lu, H.; Liu, G. Fishmeal application induces antibiotic resistance gene propagation in mariculture sediment. Environ. Sci. Technol. 2017, 51, 10850–10860. [Google Scholar] [CrossRef]
- Willems, A.; Gillis, M. Hydrogenophaga. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Yi, Y.; Zhang, Z.; Zhao, F.; Liu, H.; Yu, L.; Zha, J.; Wang, G. Probiotic potential of Bacillus velezensis JW: Antimicrobial activity against fish pathogenic bacteria and immune enhancement effects on Carassius auratus. Fish Shellfish Immunol. 2018, 78, 322–330. [Google Scholar] [CrossRef]
- Kuebutornye, F.K.A.; Abarike, E.D.; Lu, Y.; Hlordzi, V.; Sakyi, M.E.; Afriyie, G.; Wang, Z.; Li, Y.; Xie, C.X. Mechanisms and the role of probiotic Bacillus in mitigating fish pathogens in aquaculture. Fish Physiol. Biochem. 2020, 46, 819–841. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wu, Y.; Jiang, L.; Tan, A.; Zhang, R.; Luo, L. Multi-drug resistance mediated by Class 1 integrons in Aeromonas isolated from farmed freshwater animals. Front. Microbiol. 2016, 7, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igbinosa, I.H.; Igumbor, E.U.; Aghdasi, F.; Tom, M.; Okoh, A.I. Emerging Aeromonas species infections and their significance in public health. Sci. World J. 2012, 2012, 625023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adderson, E.E.; Boudreaux, J.W.; Hayden, R.T. Infections caused by coryneform bacteria in pediatric oncology patients. Pediatr. Infect. Dis. J. 2008, 27, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Evtushenko, L.I. Microbacteriaceae. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Su, H.; Hu, X.; Wang, L.; Xu, W.; Xu, Y.; Wen, G.; Li, Z.; Cao, Y. Contamination of antibiotic resistance genes (ARGs) in a typical marine aquaculture farm: Source tracking of ARGs in reared aquatic organisms. J. Environ. Sci. Health Part B 2020, 55, 220–229. [Google Scholar] [CrossRef]
- Jia, J.; Guan, Y.; Cheng, M.; Chen, H.; He, J.; Wang, S.; Wang, Z. Occurrence and distribution of antibiotics and antibiotic resistance genes in Ba River, China. Sci. Total Environ. 2018, 642, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.; Brockhurst, M.A. Plasmid-mediated horizontal gene transfer is a coevolutionary process. Trends Microbiol. 2012, 20, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.T.H.; Rossi, P.; Dinh, H.D.K.; Pham, N.T.A.; Tran, P.A.; Ho, T.T.K.M.; Dinh, Q.T.; De Alencastro, L.F. Analysis of antibiotic multi-resistant bacteria and resistance genes in the effluent of an intensive shrimp farm (Long An, Vietnam). J. Environ. Manag. 2018, 214, 149–156. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y.; Wu, J.; Luo, Y. Effects of microplastics on distribution of antibiotic resistance genes in recirculating aquaculture system. Ecotoxicol. Environ. Saf. 2019, 184, 109631. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, X.; Chen, X.; Zhao, Z.; Fang, L.; Chen, B.; Jiang, J.; Luan, T.; Chen, B. Occurrence of antibiotic resistance genes in extracellular and intracellular DNA from sediments collected from two types of aquaculture farms. Chemosphere 2019, 234, 520–527. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, X.X.; Jia, S.; Huang, K.; Tang, J.; Shi, P.; Ye, L.; Ren, H. Metagenomic insights into ultraviolet disinfection effects on antibiotic resistome in biologically treated wastewater. Water Res. 2016, 101, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; Wei, B.; OuYang, W.Y.; Huang, F.Y.; Zhao, Y.; Xu, H.J.; Zhu, Y.G. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 2015, 49, 7356–7363. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Relative abundance of ARG types (copy of ARGs/copy of 16S rRNA genes). ARG types more than 10−6 copy of ARGs/copy of 16S rRNA genes are listed, while the rest are regarded as ‘others’. (b) Percentages of different ARG types. (c) Relative abundance of ARGs detected from water/sediment in two types of shrimp ponds/corresponding downstream rivers (copy of ARGs/copy of 16S rRNA genes). The box-plot presents the biological replicates of all samples and the notch the 95% confidence intervals of the median.
Figure 1.
(a) Relative abundance of ARG types (copy of ARGs/copy of 16S rRNA genes). ARG types more than 10−6 copy of ARGs/copy of 16S rRNA genes are listed, while the rest are regarded as ‘others’. (b) Percentages of different ARG types. (c) Relative abundance of ARGs detected from water/sediment in two types of shrimp ponds/corresponding downstream rivers (copy of ARGs/copy of 16S rRNA genes). The box-plot presents the biological replicates of all samples and the notch the 95% confidence intervals of the median.

Figure 2.
Bacterial community structures of all environmental samples collected from the P. clarkii and M. rosenbergii aquaculture environments at the phylum level.
Figure 2.
Bacterial community structures of all environmental samples collected from the P. clarkii and M. rosenbergii aquaculture environments at the phylum level.

Figure 3.
(a) Network analysis revealing the correlation between bacterial community and ARGs. A connection represents a significant positive correlation (R ≥ 0.60, p ≤ 0.01) according to the Spearman’s correlation coefficient. The size of node displayed the degree of connection. (b) Relative abundance (‰) of potential pathogens in water and sediment samples in aquaculture ponds.
Figure 3.
(a) Network analysis revealing the correlation between bacterial community and ARGs. A connection represents a significant positive correlation (R ≥ 0.60, p ≤ 0.01) according to the Spearman’s correlation coefficient. The size of node displayed the degree of connection. (b) Relative abundance (‰) of potential pathogens in water and sediment samples in aquaculture ponds.

Figure 4.
(a) Relative abundance of different resistance mechanisms of ARGs in all environmental samples collected in P. clarkii and M. rosenbergii aquaculture environments. (b) Relative abundance of MGEs in water and sediment samples.
Figure 4.
(a) Relative abundance of different resistance mechanisms of ARGs in all environmental samples collected in P. clarkii and M. rosenbergii aquaculture environments. (b) Relative abundance of MGEs in water and sediment samples.

Figure 5.
Variation partitioning analysis differentiates the effects of biodiversity and MGEs on resistome alterations in aquaculture environments.
Figure 5.
Variation partitioning analysis differentiates the effects of biodiversity and MGEs on resistome alterations in aquaculture environments.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Information about the sampling sites.
| Sampling Site Sitesites | City | GPS | Size | Sampling Date |
|---|---|---|---|---|
| P. clarkii | ||||
| NJ1 | Nanjing | 32°10′56″ N 118°50′59″ E | 1.47 ha | November, 2018 |
| NJ2 | Nanjing | 32°11′7″ N 118°51′34″ E | 1.33 ha | November, 2018 |
| XY1 | Xuyi | 32°57′35″ N 118°31′2″ E | 4.00 ha | May, 2019 |
| XY2 | Xuyi | 32°45′14″ N 118°33′38″ E | 1.33 ha | May, 2019 |
| M. rosenbergii | ||||
| GY1 | Gaoyou | 32°54′32″ N 119°33′35″ E | 1.13 ha | July, 2019 |
| GY2 | Gaoyou | 32°54′30″ N 119°33′32″ E | 1.67 ha | July, 2019 |
| GY3 | Gaoyou | 32°54′30″ N 119°33′32″ E | 1.67 ha | July, 2019 |
Table 2.
Reads and α-diversity information of the water and sediment samples in two types of shrimp ponds and downstream sections of the river.
Table 2.
Reads and α-diversity information of the water and sediment samples in two types of shrimp ponds and downstream sections of the river.
| Samples | Reads | 3% Cutoff | |||
|---|---|---|---|---|---|
| Effective | Normalized | OTUs | Chao1 | Shannon | |
| P. clarkii | |||||
| PNJ1w | 74,943 | 42,536 | 5748 | 24,684.75 | 8.04 |
| PNJ2w | 75,447 | 42,536 | 5504 | 24,172.09 | 8.21 |
| PXY1w | 64,800 | 42,536 | 5375 | 22,560.42 | 8.02 |
| PXY2w | 69,032 | 42,536 | 7041 | 32,291.63 | 8.88 |
| PXYR1w | 58,791 | 42,536 | 3172 | 12,715.01 | 6.96 |
| PXYR2w | 75,569 | 42,536 | 6045 | 24,304.46 | 7.35 |
| PNJ1s | 68,900 | 42,536 | 11,747 | 32,961.82 | 11.87 |
| PNJ2s | 67,133 | 42,536 | 13,008 | 39,020.42 | 12.06 |
| PXY1s | 59,419 | 42,536 | 17,639 | 107,074.68 | 12.51 |
| PXY2s | 74,825 | 42,536 | 9752 | 33,679.55 | 10.96 |
| PXYR1s | 74,483 | 42,536 | 10,655 | 27,083.08 | 11.51 |
| PXYR2s | 70,192 | 42,536 | 7799 | 22,096.71 | 9.26 |
| M. rosenbergii | |||||
| MGY1w | 104,391 | 42,536 | 4532 | 17,284.90 | 7.48 |
| MGY2w | 102,415 | 42,536 | 4016 | 16,575.88 | 6.34 |
| MGY3w | 94,063 | 42,536 | 4796 | 20,204.21 | 7.45 |
| MGYRw | 88,096 | 42,536 | 8496 | 36,354.26 | 9.73 |
| MGY1s | 98,335 | 42,536 | 8878 | 31,662.12 | 10.45 |
| MGY2s | 93,417 | 42,536 | 9771 | 31,961.08 | 10.95 |
| MGY3s | 101,897 | 42,536 | 9618 | 35,152.96 | 10.41 |
| MGYRs | 89,043 | 42,536 | 9744 | 25,425.28 | 11.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fang, H.; Ye, N.; Huang, K.; Yu, J.; Zhang, S. Mobile Genetic Elements Drive the Antibiotic Resistome Alteration in Freshwater Shrimp Aquaculture. Water 2021, 13, 1461. https://doi.org/10.3390/w13111461
AMA Style
Fang H, Ye N, Huang K, Yu J, Zhang S. Mobile Genetic Elements Drive the Antibiotic Resistome Alteration in Freshwater Shrimp Aquaculture. Water. 2021; 13(11):1461. https://doi.org/10.3390/w13111461
Chicago/Turabian StyleFang, Hao, Nan Ye, Kailong Huang, Junnan Yu, and Shuai Zhang. 2021. "Mobile Genetic Elements Drive the Antibiotic Resistome Alteration in Freshwater Shrimp Aquaculture" Water 13, no. 11: 1461. https://doi.org/10.3390/w13111461
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.