
Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water
by
 , and
, and
Oriol Gibert
1,2,*, 
César Valderrama
1,2,
María M. Martínez
1,2,
Rosa Mari Darbra
1, 
Josep Oliva Moncunill
1 and
Vicenç Martí
1,2,3 1
Chemical Engineering Department, EEBE, Technical University of Catalonia (UPC), Av. Eduard Maristany 16, 08019 Barcelona, Spain
2
Barcelona Research Center in Multiscale Science and Engineering, EEBE, Technical University of Catalonia (UPC), Av. Eduard Maristany 16, 08019 Barcelona, Spain
3
Eurecat-Centre Tecnològic de Catalunya, Sustainability Area, Pça. de la Ciència 2, 08243 Manresa, Spain
*
Author to whom correspondence should be addressed.
Water 2021, 13(11), 1493; https://doi.org/10.3390/w13111493
Submission received: 16 April 2021
/
Revised: 13 May 2021
/
Accepted: 23 May 2021
/
Published: 27 May 2021
(This article belongs to the Special Issue Wastewater Treatment by Adsorption and/or Ion-Exchange Processes for Resource Recovery)
Abstract
:An approach for the remediation of heavy metal-contaminated wastewater that is gaining increasing attention is the application of hydroxyapatite (HAP)-based particles. HAP is conventionally synthesized through wet chemical precipitation of calcium and phosphate ions, although later studies have focused on HAP synthesis from solid calcite contacted with a phosphate solution under ambient conditions. This synthesis route can allow saving soluble Ca-chemicals and, thus, make the process more cost-efficient. The aim of this study was to coat natural calcite powder with a layer of HAP for the removal of Zn and Cu from contaminated water. For this purpose, a HAP layer was synthesized on calcite particles, characterized using several complementary techniques and evaluated for the removal of Zn and Cu from synthetic solutions. Sorption kinetics and equilibrium isotherms, as well as the effect of sonication of the synthesized sample on its sorption performance, were determined. The results showed that calcite particles were efficiently coated with a HAP layer with high capacity in removing Zn and Cu from acidic solutions, with a qmax of 34.97 mg/g for Zn (increased to 37.88 g/mg after sonication of the sample) and 60.24 mg/g for Cu (which hardly varied with sonication). The mechanisms behind the sorption of Zn and Cu onto HAP, inferred from pH changes, the relation between metal uptake and Ca2+ release and XRD analysis, included surface complexation, ion exchange and precipitation of new Zn- and Cu-containing phases.
1. Introduction
Acidic metal-rich wastewater from mining and industrial processes represents an environmental problem worldwide due to its low pH and high content of heavy metals. It is well-known that heavy metals discharged into the environment may pose a serious hazard to all living beings because of their bioaccumulation and toxicity even at low concentrations [1,2]. Therefore, removal of heavy metals from this type of wastewater by an appropriate treatment becomes necessary.
When wastewater can be managed on the surface, above-ground engineered systems offer the opportunity to customize the operation conditions for an optimized performance. The removal of heavy metals in these systems can be achieved through several processes, with the most common ones being chemical precipitation, coagulation-flocculation, membrane filtration, ion-exchange, electrochemical processes and sorption. Chemical precipitation is the simplest and most employed approach, although it consumes large amounts of chemicals and can generate an excessive amount of sludge that requires further treatment [3,4]. Coagulation-flocculation also removes heavy metals from water with reduced time to settle suspended solids, but like chemical filtration, it produces huge amounts of sludge that need to be handled and treated [3,5]. Membrane filtration (particularly nanofiltration and reverse osmosis) is recognized as a very efficient technology for the removal of dissolved heavy metals, although it usually requires high energy consumption and suffers from membrane fouling, posing difficulties in their operation and increasing operation costs [6,7]. Ion-exchange has also proved to be useful for the removal and recovery of heavy metals from water. It offers the advantages of selective removal and no sludge production, but on the other hand, ion-exchange resins are expensive and their regeneration can increase the cost even further. Moreover, successful removal by ion-exchange is applicable to only a limited number of metal cations [5,8]. Electrochemical methods are also an option to remove heavy metals from water, but they are usually associated to high costs and high electrical supply [3,7]. Finally, sorption arises as one of the most attractive methods thanks to its efficiency, versatility, simplicity in design and operation and low cost [5,9]. Sorption-based approaches are spreading as new sorbents and applications are researched. Within this context, the recent developments in micro- and nano-particles (MPs and NPs) with sorption capacity towards metals are gaining increased attention of researchers [2,10].
When contaminated water is groundwater (e.g., when mining acidic metal-rich streams known as acid mine drainage (AMD) percolate through the soil), the selection of the most suitable technology becomes more complicated. First, some of the above-mentioned processes (e.g., membrane filtration or ion-exchange) are technically impractical at depth and/or are cost-prohibitive when applied on surface after groundwater extraction. In these cases, in-situ remediation in the aquifer itself through the installation of a permeable reactive barrier (PRB) [11], the insertion into the subsoil of electrodes to promote electrochemical processes [12] or the introduction of reactive chemicals via injection wells [13] has become a matter of active research. Despite successful experiences, PRBs have shown to be restricted to shallow aquifers and often suffer from limited capture of the contaminated plume [11,14]. For their part, technologies based on in-situ-induced electrochemical processes are constrained by the limited metal removal and high energy consumption [12]. Another promising approach for in-situ remediation that is gaining increasing attention of researchers is the application of MPs and NPs in the subsoil able to immobilize, neutralize or decompose the contaminants present in groundwater, as the application of MPs and NPs allows reaching contaminants in deep aquifers not accessible by PRBs [10,15].
Many different materials have been evaluated for their application in the form of MPs and NPs [16,17]. For the removal of heavy metals, powders based on hydroxyapatite (Ca10(PO4)6(OH)2, hereafter referred to as HAP) have been acknowledged as one of the most suitable reactants thanks to its high sorption capacity for heavy metals, low water solubility, availability and low cost, environmental compatibility and high stability under oxidizing and reducing conditions [18,19,20].
Properties (and thus performance) of a given HAP greatly depend on how it has been synthesized [21,22]. Among the conventional methods of producing HAP particles, the classical wet chemical precipitation (i.e., soluble Ca2+ and HPO42− are precipitated in an aqueous solution at basic conditions) is often reported to be the most convenient one because, unlike more sophisticated strategies, it is not associated to high cost and energy use [23,24]. The final composition, morphology and properties of the synthesized HAP following the wet precipitation method depend on many variables, such as concentration and proportion of precursors, pH, temperature, presence of other ionic species or organic compounds and aging time [21,25,26]. Numerous studies have demonstrated that HAPs with high sorption capacity for metals can be synthesized over a wide range of values for these variables.
Modifications of the classical wet chemical precipitation of HAP have been investigated in other fields. HAP has been synthesized, under ambient conditions, from solid calcite contacted with a phosphate solution, on the basis that the solid calcite acts as a source of Ca2+ instead of externally adding it in the form of soluble Ca2+. Under this scheme, Ca2+ is supplied by dissolution of CaCO3 and precipitated by the following overall reaction:
10CaCO3(s) + 6HPO42− + 4H2O → Ca10(PO4)6(OH)2 + 10HCO3−+ 2OH−
This approach has been applied for the synthesis of HAP from biogenic calcite (e.g., nacre, oyster shells, sea urchin spines or other natural osteo-inductive biomaterials) [20,26,27,28]. Further studies have compared the effect of using different orthophosphate sources in such a synthesis route [29]. In addition, under this scheme, studies aiming at protecting marble sculptures and monuments from acid rain have investigated the possibility of coating them with a film of HAP synthesized by using the marble itself as a Ca2+ source for the HAP [30,31,32]. Although these studies provide very promising results, further investigation is needed to fully understand HAP synthesis from solid CaCO3.
From a remediation perspective, preparing calcite particles coated by HAP for sorbing metals would allow saving huge amounts of soluble Ca-chemicals and, thus, make the process more cost-efficient and improve its competitiveness. Solid calcite, as one of the fifth most abundant biominerals in the Earth crust, has a very low cost and wide availability. However, to the best of our knowledge, there is a lack of studies from such a standpoint and only an attempt has been made to synthesize phosphatized dolomite as sorbent to be used for the treatment of metal-contaminated water [33].
Within this context, the aim of this bench-scale study was to coat natural calcite powders with a layer of HAP for the removal of heavy metal ions (Cu and Zn) from contaminated water. Zn and Cu ions were chosen as representative heavy metals, with wide presence in the environment caused mainly by human activity. The specific objectives were (1) to synthesize and characterize a layer of HAP on solid calcite particles, (2) to assess its sorption capacity toward Zn and Cu in single-metal solutions, determining the sorption kinetics and the equilibrium isotherms (and to compare them with those of commercial HAPs), (3) to evaluate the effect of sonicating the material on sorption performance and (4) to infer the mechanisms responsible for the removal of Zn and Cu.
2. Materials and Methods
2.1. Bare Calcite MPs and Chemical Reagents
The commercial calcite MPs were purchased from Reverté S.A. (Spain). The MPs were found to have an average particle size of 10 μm and exhibit a BET surface area of 0.87 m2/g. The acid digestion of calcite MPs and analysis by ICP-OES showed a composition of 36.5% Ca and 0.17% Mg. Precursors (NH4)2HPO4 (Scharlau ≥ 98%), CaCl2 (Panreac ≥ 98%), NaOH (Panreac ≥ 98%) and ethanol (Scharlau ≥ 96%) were used for HAP synthesis. For sorption experiments, acidic Zn and Cu solutions were prepared from their ZnCl2 and CuSO4 salts (Panreac ≥ 98%) and HNO3 (Panreac ≥ 96%). Performance of synthesized HAP was compared against commercial micro-HAP (Acros 35–40% in Ca) and nano-HAP (Merck ≥ 97%), labeled hereafter as mHAPcom and nHAPcom, respectively.
2.2. Synthesis Route of HAP Coatings on Calcite MPs
The synthesis route of the HAP coating consisted of a two-step procedure based on that applied by Graziani et al. for the coverage of marble with a HAP film [32]. First, 1 g of calcite MPs was placed in a plastic centrifuge tube and put into contact with 25 mL of a solution containing (NH4)2HPO4 (0.1 M), CaCl2 (0.1 M) and a pH of 8 in the presence of ethanol 0.5%, as ethanol has been found to improve the coverage of a calcite surface by a more uniform, crack-free and thinner layer of HAP [32]. The pH was adjusted by adding NaOH dropwise until the desired value was achieved. Second, a further precipitation of HAP following the same procedure and concentrations as described above but without ethanol was carried out.
For each step, the suspension was shaken in a rotary shaker (Heidolph GmbH, REAX 20, Schwabach, Germany) for 24 h. This time was chosen to allow possible precipitated intermediates (octacalcium phosphate, amorphous calcium phosphate, calcium-deficient hydroxyapatite) to shift to HAP [23]. After 24 h, the synthesized solid (HAP-coated calcite MPs) was separated by centrifugation (J.P. Selecta, Centronic-BLT, Abrera, Spain) at 4000 rpm for 20 min, oven-dried (J.P. Selecta, Digiheat, Abrera, Spain) at 110 °C for 1.5 h and ground with a mortar before further use. This material was labelled “synt” (from synthesized).
In order to diminish the particle size of the selected material and thus potentially enhance its sorption capacity, a portion of it was subjected to sonication, as it is known that ultrasonic energy applied to a suspension can lead to breakage of agglomerated particles. Sonication of a sample of 4 g in 250 mL of deionized water was conducted in an ultrasonic cleaner (ATU Ultrasonidos, ATM40-2L-CD, Paterna, Spain) operating at a fixed frequency of 40 Hz for 30 min. The solid was then separated by centrifugation and dried as described above. The modified material was labelled “son” (from sonicated).
Performance of samples “synt” and “son” were compared against that of commercial HAP samples mHAPcom (micro-size) and nHAPcom (nano-size).
2.3. Characterization of the HAP-Coated MPs Samples
The synthesized material and its sonicated sub-sample were characterized using several complementary techniques. Functional groups were characterized employing a Fourier Transform Infrared (FTIR) spectrophotometer (Nicolet 6700, Madison, WI, USA) fitted with an Attenuated Total Reflectance (ATR) accessory (Smart Orbit). Mineralogical identity and crystallinity were analyzed by X-ray powder diffraction (XRD) recorded on a Bruker D8 Advance diffractometer. Morphology was viewed with a Focused Ion Beam–Scanning Electron Microscope (FIB-SEM) equipped with an energy dispersive spectrometer (EDS) for elemental analysis (Carl Zeiss Neon40, Jena, Germany). Samples were mounted on stainless-steel stubs using a double-sided adhesive carbon disc and sputter-coated with a thin layer of carbon to render them conductive for SEM observation. The specific BET surface area (SBET) of the samples was measured by N2 adsorption according to the Brunauer–Emmet–Teller method in a SBET analyzer (Micromeritics ASAP 2020, Aachen, Germany). Degassing and heating was carried out at a maximum temperature of 250 °C. Particle size distribution (PSD) was determined using a Laser Diffraction Particle Size analyzer (Malvern Panalyitical, MasterSizer 3000, Worcestershire, UK) with a Hydro EV wet dispersion unit. For calculations, the Mie theory was chosen, and refractive and absorption indices were 1.630 and 0.010, respectively. In order to obtain PSD of particles in the same conditions of the experiments, measurements were carried out with water and without application of ultrasounds. PSD curves provided by the instrument were given as volumetric PSD. The pH at the point zero charge (pHPZC) was estimated through the immersion technique by putting an amount of solid material in contact with solutions at different initial pHs and measuring the change of pH (ΔpH) once equilibrium was reached [34].
2.4. Sorption Performance of the Synthesized HAP-Coated Material
2.4.1. General Procedure
All sorption experiments were carried out using standard batch methodology. Known volumes (50 mL) of prepared solutions of Zn and Cu (added in the form of ZnCl2 and CuSO4) at pH 4.6 were contacted with a weighted amount of sorbent in plastic centrifuge tubes and shaken in a rotatory shaker for 24 h at room temperature (21 ± 2 °C). For the kinetics study, the weighted amount was 0.05 mg, the Zn or Cu concentration was 50 mg/L and the supernatant solution samples were collected at predetermined time intervals (after 1, 2, 4, 6, 9, 12, 15, 25, 40, 60 min). For the equilibrium experiments, the weighted amounts of sorbent were 0.05 or 0.1 g, and the initial Zn or Cu concentrations were 5, 10, 20, 40, 60, 80, 100 and 120 mg/L. The ratio of aqueous phase volume (mL) to sorbent weight (mg) was varied as necessary to observe a measurable decrease in the concentration of Zn and Cu in the aqueous solution once the equilibrium was reached. No pH control was imposed during sorption experiments in order to mimic the subsoil conditions during the remediation of contaminated groundwater. All centrifuge tubes were shaken in a rotary shaker at 50 rpm for 24 h, after which the supernatant solution was separated from the sorbent by centrifugation at 4000 rpm for 10 min, filtered (0.22 μm), acidified (1% HNO3) and transferred to auto-sampler vials for analysis of the remaining Zn and Cu. The ions Ca2+, HPO42− and pH in the supernatant were also determined in separate aliquots.
2.4.2. Sorption Kinetics of Zn and Cu
The kinetics of the sorption of Zn and Cu onto the HAP was fitted using the pseudo first-order and the pseudo second-order kinetics models, whose linearized forms are, respectively:
where q and qe are the amounts of adsorbed metal per weight unit of adsorbent (mg/g) at equilibrium and at time t respectively, and k1 and k2 are the rate constants of pseudo first-order (min−1) and pseudo second-order (g·mg−1·min−1), respectively.
The amount of adsorbed metal per weight unit of the adsorbent (q, in mg/g) was calculated from the following mass balance, also valid for the equilibrium:
where ci and ce are the initial and equilibrium concentrations of the metal ion in the bulk solution (mg/L), V is the volume of the solution (L) and w is the weight of the sorbent (g).
2.4.3. Sorption Isotherms for Single-Metal Systems
Sorption equilibrium data are usually plotted as an isotherm displaying the content of the sorbed species on the sorbent (qe) (in mg/g) versus the concentration of the remaining aqueous species (ce) (in mg/L). The sorption data of this study were modeled taking into account the concentration of ions in both phases and according to the widely used Langmuir equations:
where qmax is the maximum adsorption capacity (mg/g) and KL is the Langmuir binding constant, which is related to the energy of adsorption (L/mg). Experimental ce and qe data were used to determine the constants (qmax, KL) for each metal and sorbent from the linearized form of the Langmuir isotherm and using least-square regression analysis.
2.5. Water Analysis
Measurements of pH were made with a Hamilton combination pH electrode coupled to a Crison GLP22 pH meter. HPO42− and Ca2+ in the supranatants in the sorption experiments were analyzed by ionic chromatography (IC) (Dionex, ICS-1000, Sunny Valley, ID, USA) equipped with cationic and anionic detectors (ICS-1000 and ICS-1100, respectively) and controlled by software Chromeleon®chromatographic. Ca, P and Mg (in the determination of the calcite composition), and Zn and Cu (in the sorption experiments) were analyzed by atomic absorption spectroscopy (AAS) (Varian, SpectraAA 50-B, Palo Alto, CA, USA) and inductively coupled plasma optical emission spectrometry (ICP-OES) (Perkin Elmer, Optima 8300, Waltham, MA, USA).
3. Results and Discussion
3.1. Characterization of the Synthesized Material
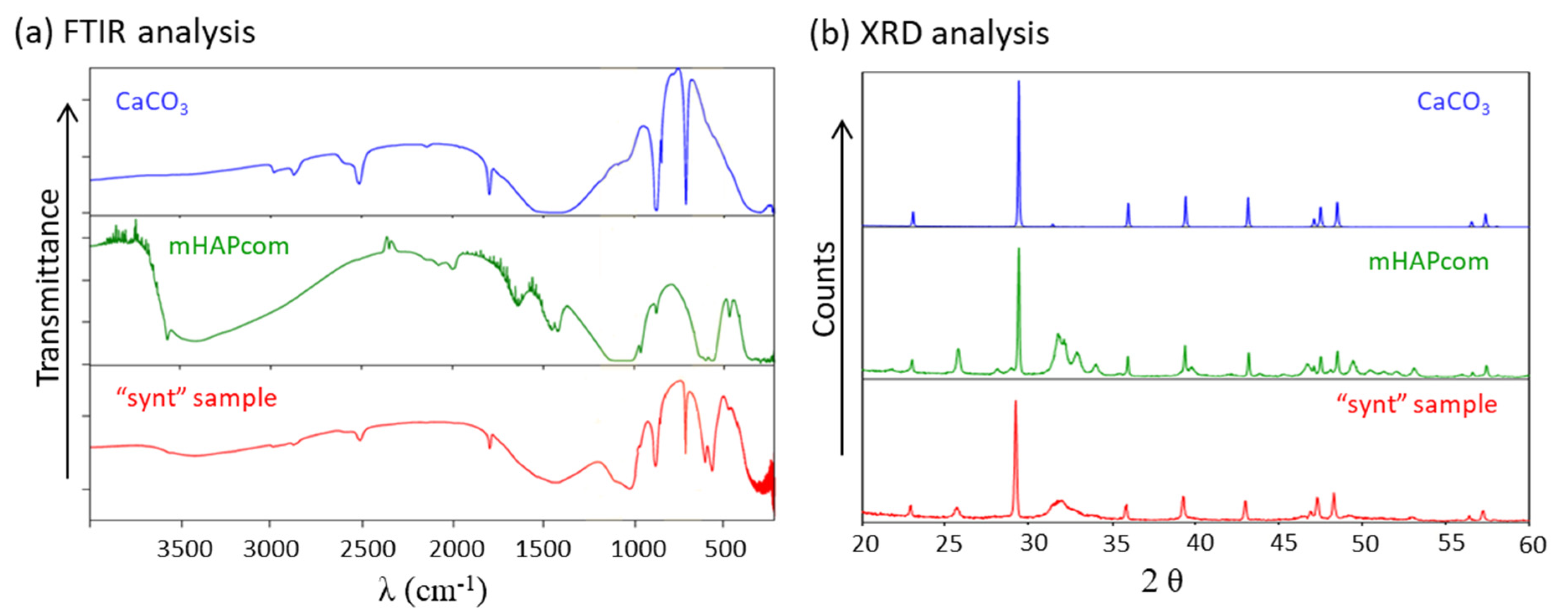
Figure 1 displays the FTIR spectra (a) and the XRD spectra (b) of the “synt” sample and, for comparison, calcite and a commercial HAP (mHAPcom).
The spectrum of the “synt” sample presented characteristic peaks and bands of HAP: a broad band centred at around 1030 cm−1 (corresponding to asymmetric stretching mode ν3 of the PO43− group) and sharp peaks at 600 and 565 cm−1 (ascribed to bending mode ν4 of the PO43− group). The slight shoulder observed at 964 cm−1 may also be assigned to symmetric stretching mode ν1 of the PO43− group, as well as the broad band observed at 3420 cm−1 which can be ascribed to the stretching vibrations ν(O-H) of the HAP (although it may also be of interstitial water) [35,36,37,38]. On the other hand, the characteristic peaks of CaCO3 were also observed: a broad band centred at about 1440 cm−1 (asymmetric stretching ν3 of CO32−) and well-resolved peaks at 880 cm−1 (out-of-plane bending ν2 of CO32−) and 715 cm−1 (symmetric in-plane bending ν4 of CO32−). An additional slight peak at 1810 cm−1 was also observed coming from stretching vibration ν(C=O), while the slight shoulder at 1060 cm−1 can be ascribed to symmetrical stretching ν(C–O) [37,39]. The FTIR spectrum for the “son” sample resembled that of the “synt” sample (not shown).
The XRD spectrum of the “synt” sample presented characteristic diffraction peaks of HAP (e.g., 25.8° and 31.8°) together with those of its precursor CaCO3 [26,31,40]. Again, the XRD spectrum for the “son” sample looked like that of the “synt” sample (not shown).
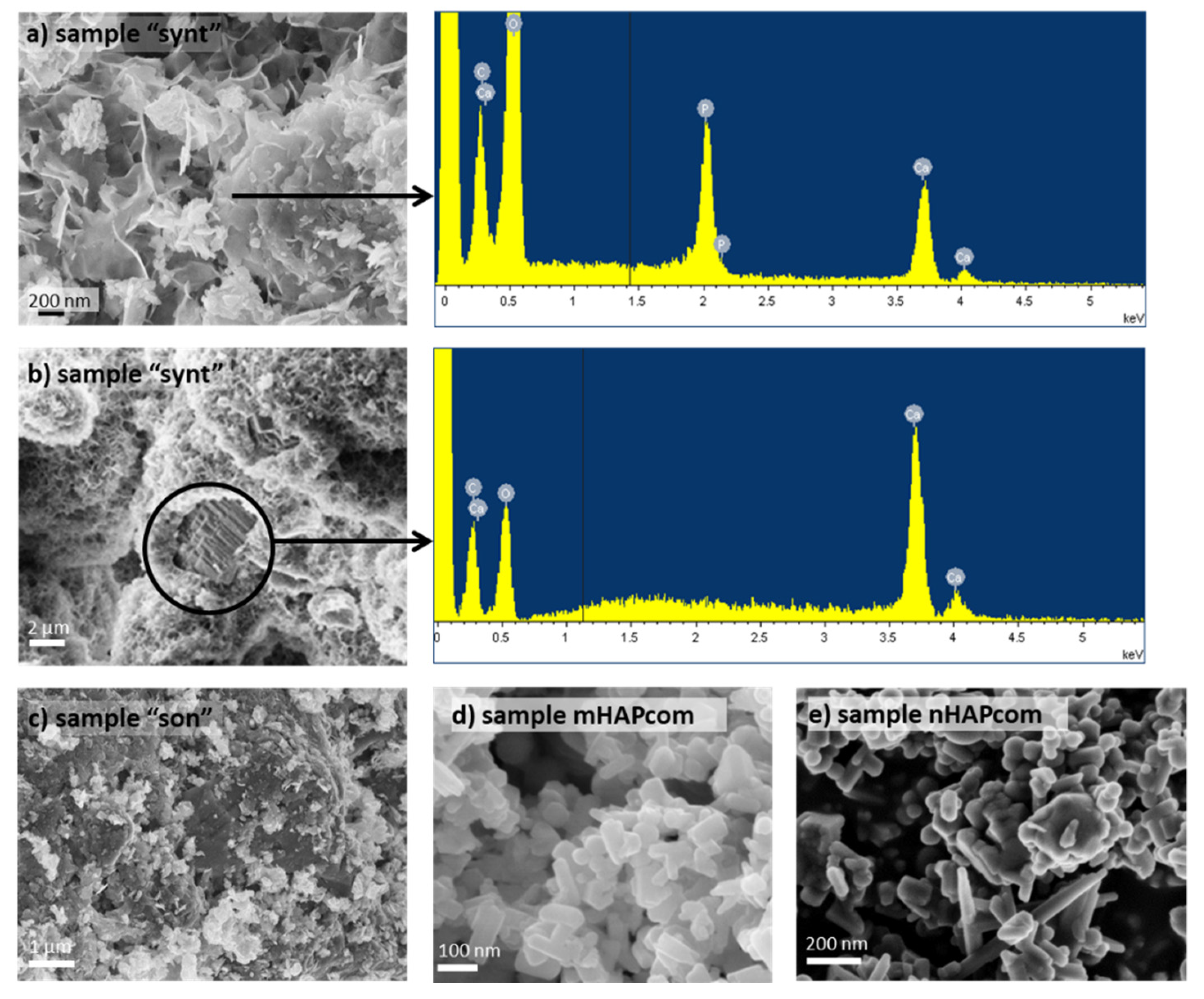
Figure 2 shows the SEM images of the sorbents used in sorption tests. SEM images of the “synt” sample showed that the surface of the calcite MPs was mostly covered by HAP, easily recognizable by its flower-like structure (Figure 2a) [41]. Consistently, EDS analysis confirmed that this layer was constituted by Ca, P and O. The layer visible by SEM did not display recognizable individual particles but a continuous coating of aggregated particles instead, making it difficult to assess an average particle size. Very occasional uncoated areas were also observed, exhibiting underlying calcite (with its characteristic layered terraces), as confirmed by EDS analysis (Figure 2b). These bare areas of calcite would help explain the loss of mass (10%) observed after the acid attack.
SEM images of the “son” sample seemed to indicate that sonication led to a detachment of HAP, yielding a surface with more bare areas of calcite and more cavities (Figure 2c). The “son” sample also exhibited a highly agglomerated structure. The associated EDS spectrum (not shown) resembled that of the uncovered areas of the “synt” sample (Figure 2b).
SEM images of the commercial macro- and nano-HAP (mHAPcom and nHAPcom, respectively) showed agglomerates of more rounded, smooth HAP particles with different size (of approximately 40 to 60 μm) (Figure 2d,e, respectively).
BET results for all studied sorbents are summarized in Table 1. It is worth noting that a notorious specific surface area increment was obtained during the synthesis process from calcite to HAP. Measured values of SBET surface areas for “synt” and “son” samples were 58.25 and 53.68 m2/g, which were similar or slightly lower than that for the mHAPcom (60.77 m2/g) and clearly higher than that for nHAPcom (18.93 m2/g). The low value of the latter was probably related to its high crystallinity. The pore volumes of the synthesized HAPs were 0.0016 cm3/g for the “synt” sample and 0.0013 cm3/g for the “son” sample.
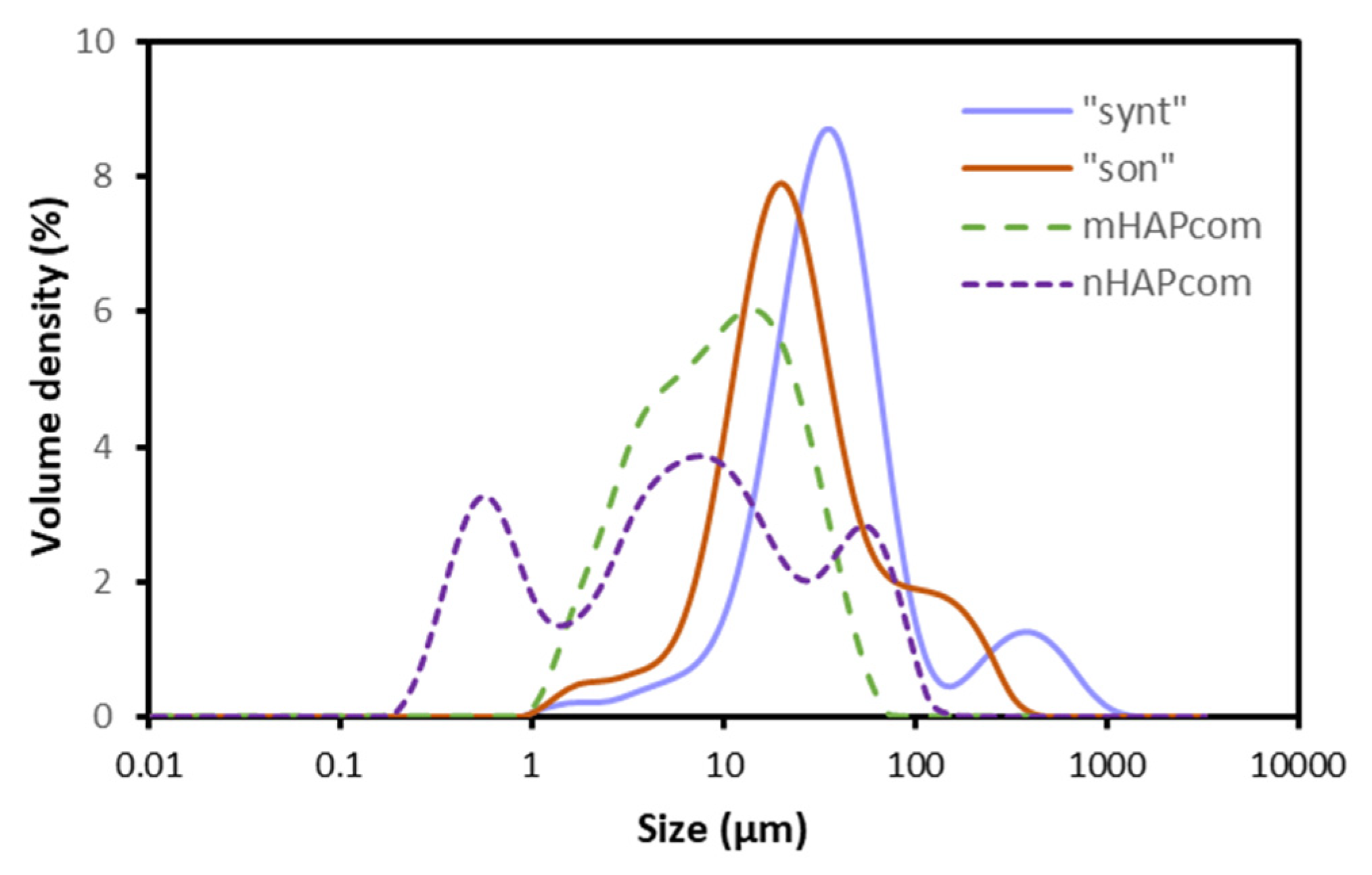
Volumetric particle size distributions (PSD) of the studied samples are represented in Figure 3. The PSD curve of “synt” and “son” samples exhibited a similar symmetric single peak with distribution widths close to each other, with a shoulder or additional small peak on the right, suggesting that agglomeration of particles took place. However, the two PSDs differed in their mean diameter: 36 μm for “synt” and 23 μm for “son”. This seemed to prove that sonication resulted to some extent in a fracturing or breakage of the agglomerates. For mHAPcom, the PSD curve revealed a mean diameter of 10 μm, which was apparently smaller than that visualized in SEM analysis (Figure 2d). For nHAPcom, the PSD curve was non-uniform, which indicated that the sample contained particles with very different size (mean diameter of 6 μm). Differences in size between PSD and SEM measurements are usually attributed to agglomeration of particles or inaccuracies in the assumption of sphericity of particles measured in PSD.
The HAP particles synthesized in this study were hence presented as agglomerates of calcite cores coated with HAP of average size of 36 μm, with a large SBET (58.25 m2/g). Sonication of this sample caused some detachment of the HAP layer, resulting in slightly smaller agglomerates (of average size of 23 μm) and SBET (53.68 m2/g).
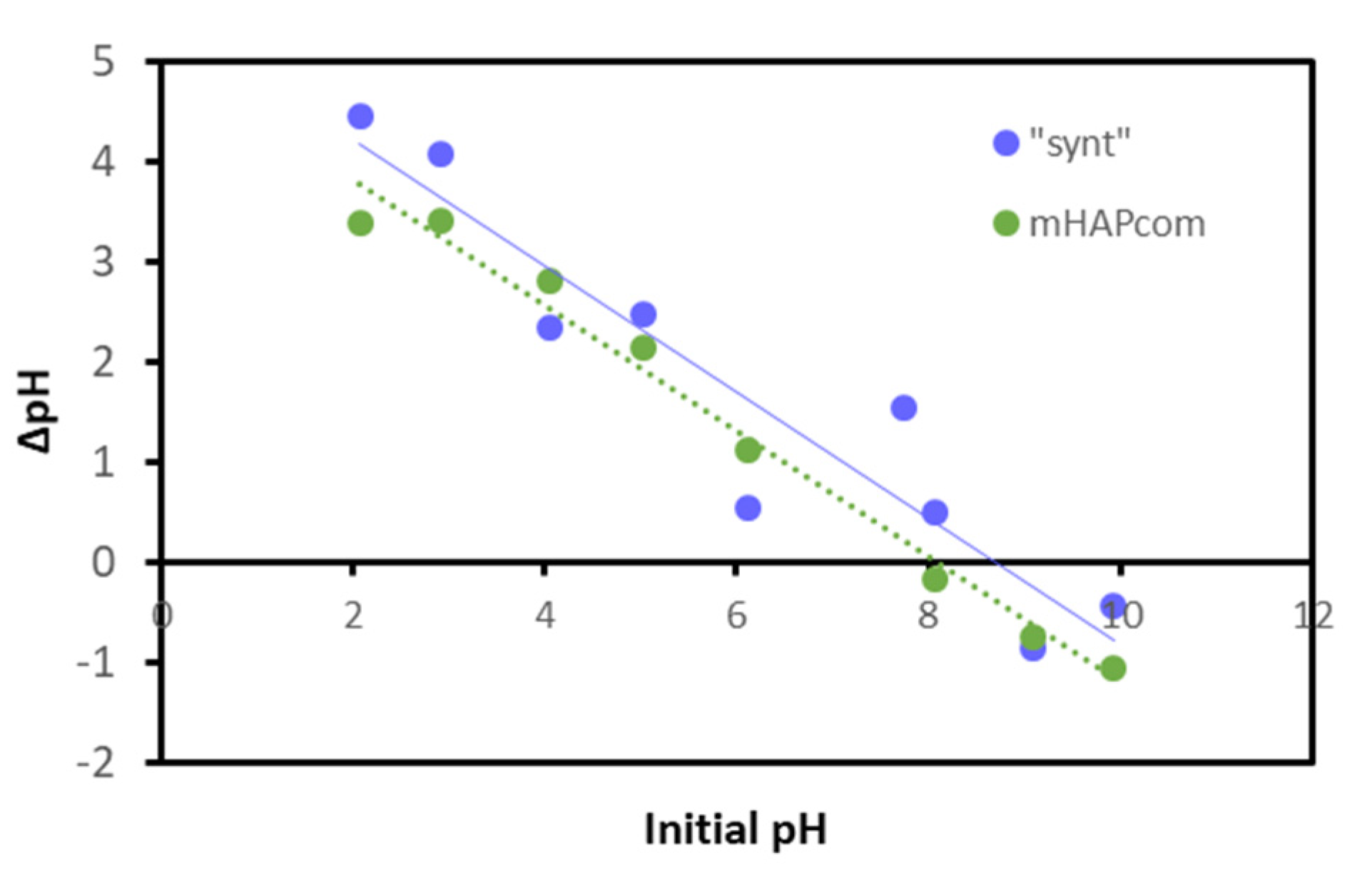
The pHPZC was estimated by the immersion technique [34]. Figure 4 represents the change of pH (ΔpH) versus the initial pH. The pHPZC was identified as the pH with no variation of pH (ΔpH = 0) and quantified to be 7.7 for the “synt” sample and 8.2 for the mHAPcom sample. These values were comparable to reported pHPZC for synthesized HAP (7.3–8.6) [25,41].
3.2. Adsorption Kinetics
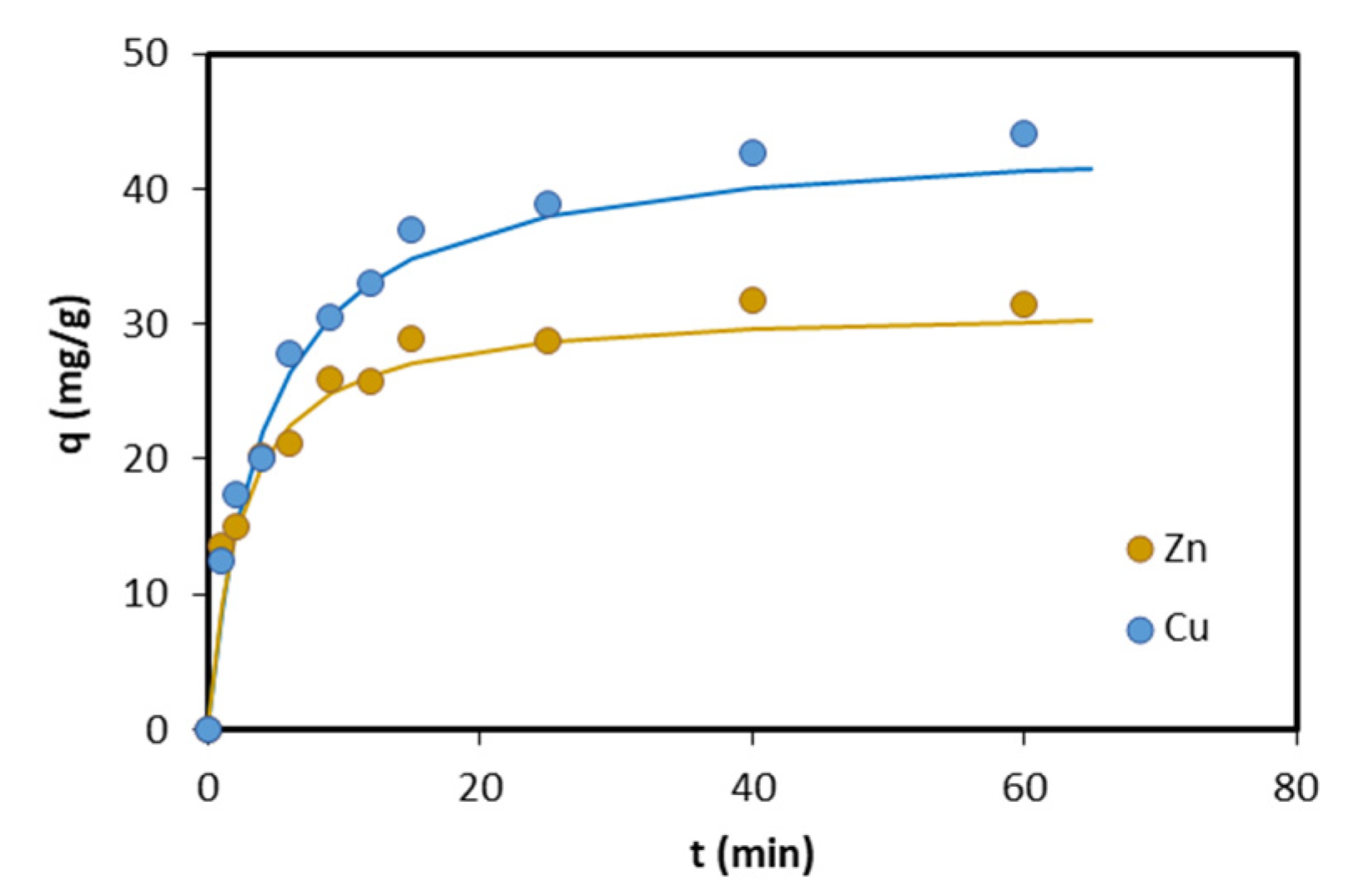
Sorption of Zn and Cu onto the “synt” sample as a function of time is illustrated in Figure 5. It can be seen that, for both metals, equilibrium between the two phases was attained in approximately 40 min.
The experimental kinetic data were fitted with pseudo-first and pseudo-second order kinetic equations. The rate constants k1 and k2, and qe values were calculated from the interception and slope of the line obtained by plotting log(qe − q) and t/q against t. The obtained values, together with regression coefficients, are shown in Table 2. The higher regression coefficients for the pseudo-second order kinetic model revealed that this model better described the kinetics of Zn and Cu sorption on HAP. The k2 values were found in the range of published values for Zn (0.001–0.88 g/(mg·min)) [25,42] and Cu (0.001–0.076 g/(mg·min)) [36,42,43,44].
3.3. Adsorption Isotherms for Single-Metal Solutions
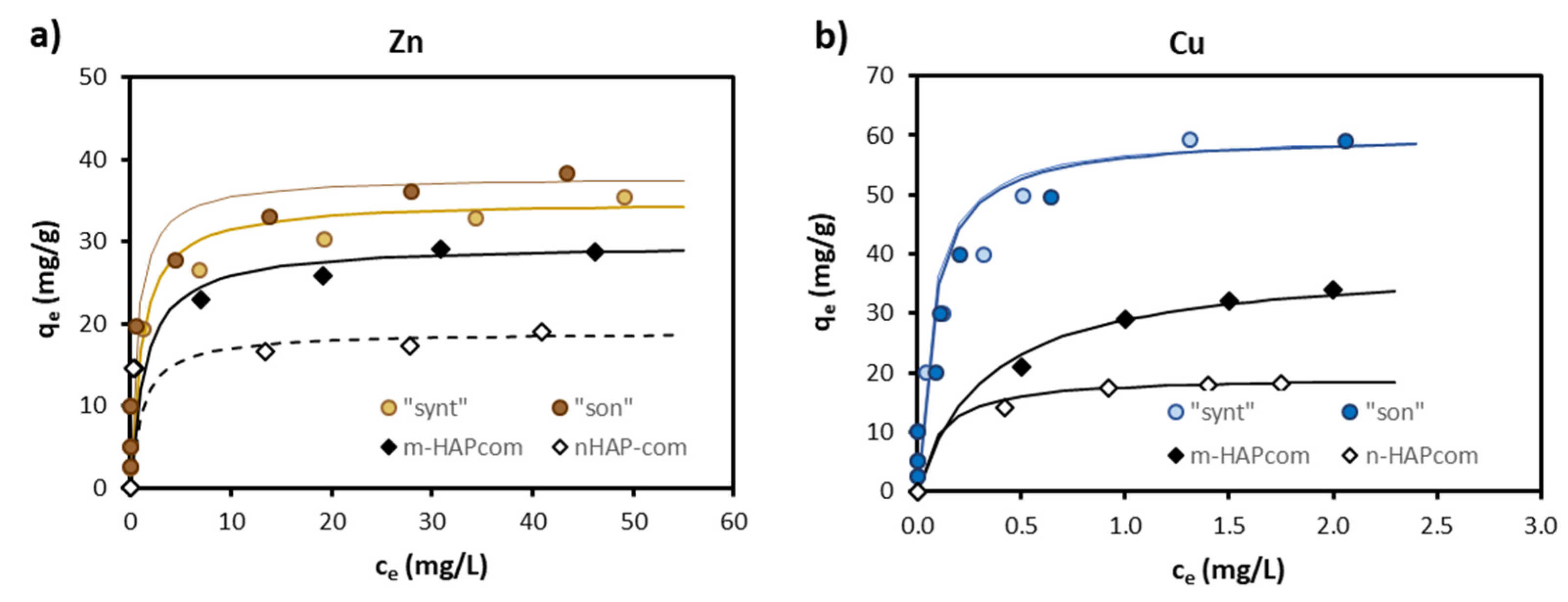
Figure 6 shows the experimental equilibrium data for the sorption of Zn and Cu in single-metal solutions onto “synt”, “son”, mHAPcom and nHAPcom materials. The fitting of the experimental ce and qe data to the Langmuir-type model gave rise to the values of the constants (qmax and KL) provided in Table 3. The Langmuir curves generated with the obtained constants are plotted in Figure 6.
The “synt” and “son” materials were efficient in sorbing Zn (qmax = 34.97–37.88 mg/g) and Cu (qmax = 60.24 mg/g), with sorption capacities higher than those of commercial HAPs (Figure 5) (note that isotherms for Cu on both “synt” and “son” sorbents in Figure 6b superpose in the whole concentration range). Considering that sorption is a surficial process, qmax were SBET-normalized. As can be seen in Table 3, the obtained values were still higher for the synthesized materials (excepting for Zn on nHAPcom).
Table 4 compares qmax obtained in this study with reported values in the literature. It must be kept in mind that sorption capacity of a given HAP largely depends on the conditions of its preparation and sorption experiments’ conditions. Therefore, Table 4 only compares studies on HAP synthesized through wet chemical precipitation and evaluated in batch sorption tests. As can be seen, values of qmax in this study were comparable to published values. In general, more crystalline HAP yielded lower qmax but also lower SBET, so that SBET-normalized qmax remained almost constant, as also observed by Šljivić et al. [43].
Some authors have explained the difference in selectivity of HAP toward different metal cations on the basis of their hydrated ion radius and electronegativity [25]. The higher hydrated ion radius (4.30 Å) and lower electronegativity (1.60) of Zn than of Cu (4.19 and 1.90 Å, respectively) and their sorption extent observed in the present study seemed to support this theory.
3.4. Sorption Mechanisms
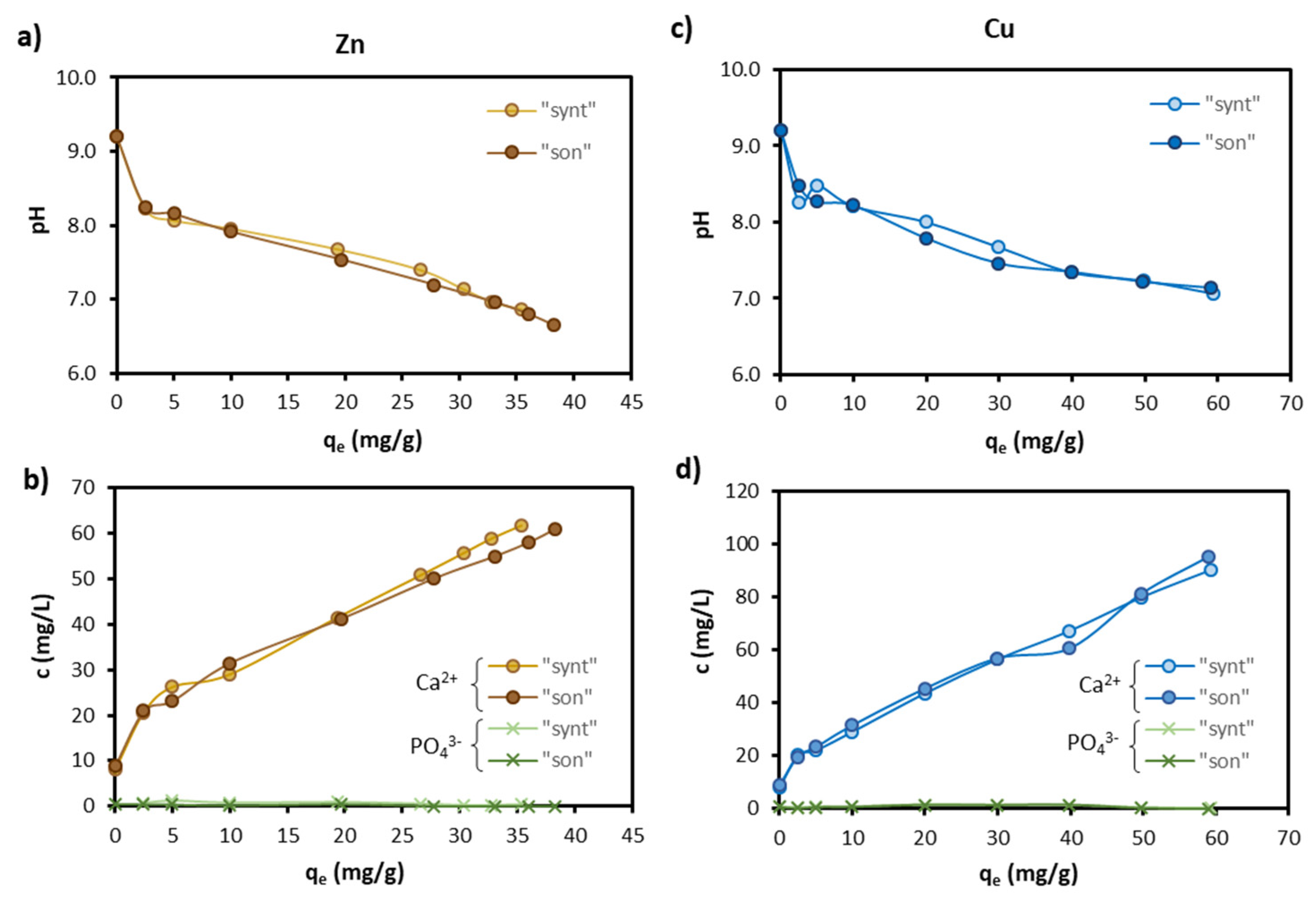
Figure 7 shows that Zn and Cu sorption was accompanied by a pH decrease (Figure 7a,c) and an increase of Ca2+ concentration in the solution (Figure 7b,d). Both were proportional to the amount of Zn and Cu sorbed onto HAP (qe).
The pH decrease can be explained by the surface complexation reaction between Zn and Cu and deprotonated surface hydroxyls of HAP, summarized as follows [42,47]:
where M2+ stands for Zn2+ or Cu2+ ions. Decreases of pH by metal sorption on HAP have also been observed by other investigators [18,25,35,43]. In the absence of M2+, the pH in the present study increased from the initial pH 4.7 to 9.2. Unlike other studies, this increase was beyond pHPZC of HAP (in the range 6.1–8.3) [18,36,43], probably because calcite in bare areas of the HAP-coated calcite MPs dissolved and caused pH to rise to higher values than HAP alone would do. It is worth noting that as long as pH remained above pHPZC (i.e., the HAP surface exhibited a negative charge), electrostatic forces between the HAP surface and the metal cation favored cation sorption [48].
≡HAP-OH + M2+ ⇆ ≡HAP-O-M+ + H+
2 ≡HAP-OH + M2+ ⇆ (≡HAP-O)2M + 2H+
2 ≡HAP-OH + M2+ ⇆ (≡HAP-O)2M + 2H+
However, the simple surface complexation process would not cause any Ca2+ concentration. The increase in Ca2+ concentration was fairly well-explained by an ion exchange process, whereby M2+ replaces a surface Ca2+ of the HAP, presented as follows:
≡HAP-Ca2+ + M2+ ⇆ ≡HAP-M2+ + Ca2+
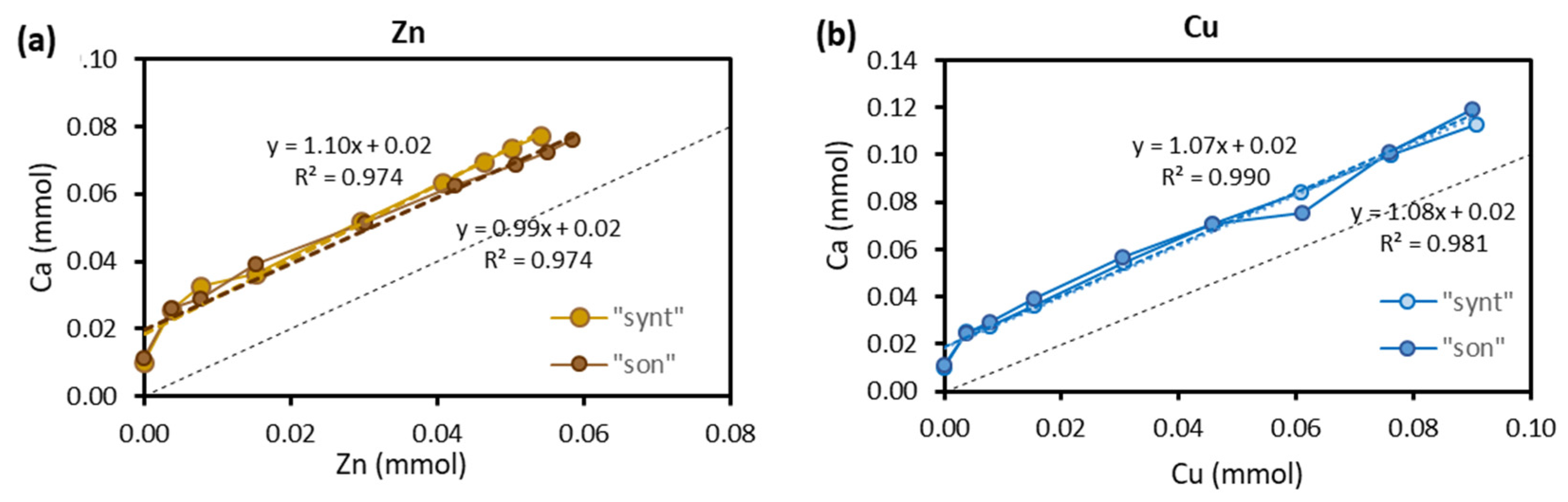
Figure 8 presents the relationship between the number of moles of released Ca2+ versus the number of moles of adsorbed Zn (Figure 8a) and Cu (Figure 8b). The relationship is linear with a slope very close to unity. A slope close to unity would suggest that Zn and Cu were removed solely by ion exchange.
Nevertheless, the decrease of pH seen above revealed that ion exchange could not be the unique mechanism behind the metal removal. Therefore, other mechanisms also took place besides ion exchange that explained the Ca2+ release. Ca2+ release might also be due to exposed calcite dissolution, which was favored as pH decreased (from 9.2 to 7.0) as metal sorbed onto HAP (Figure 7). Ca concentration value on the y-axis (i.e., in the absence of metal sorption) would support this hypothesis. Another source that might contribute to the Ca2+ release (although to a limited extent) was HAP dissolution. Crystalline HAP dissolution is reported to occur generally at pH < 5, but non-crystalline and substituted with Ca2+ HAP (as it might be the case in this study as HAP was synthesized from calcite dissolution) may dissolve at higher pH [30,49]. Ca2+ concentrations in equilibrium with HAP have been reported to range from 1.4 mg/L for highly crystalline HAP to 7.5 mg/L for poorly crystalline HAP [49]. That being the case, the insignificant concentration of PO43− (Figure 7) would indicate precipitation of PO43−-bearing solid phases. Metal precipitation with PO43– released from HAP dissolution has been observed [18,42,50].
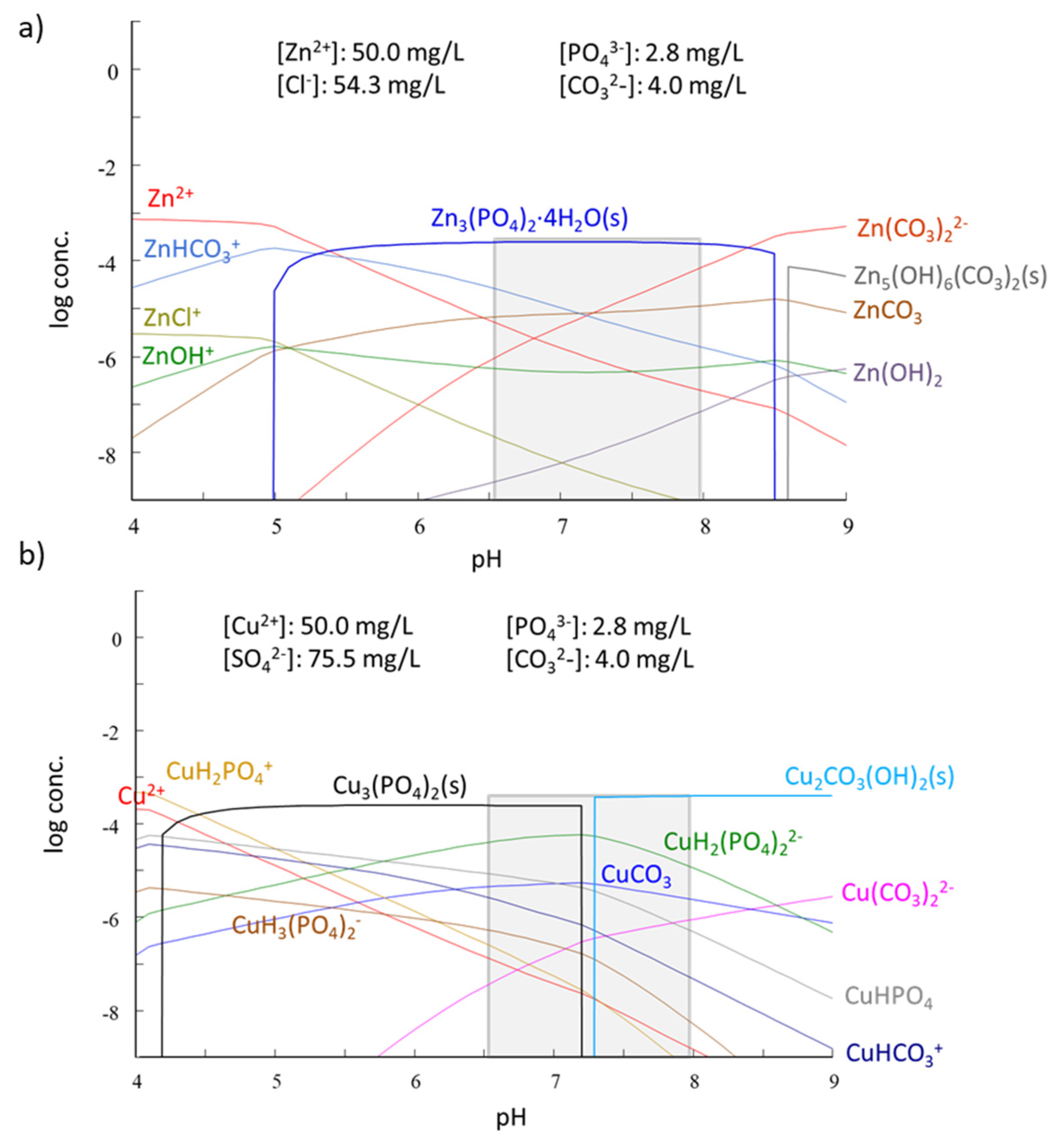
Precipitation, and not only of PO43−-bearing solid phases, is in fact a third mechanism that is expected to contribute to the overall removal of Zn and Cu. Speciation diagrams based on thermodynamic equilibria in Zn and Cu systems at pH 6.5–8 in the presence of CO32− (from calcite) and PO43− (from HAP) anticipate the formation of non-soluble phases such as Zn3(PO4)2·4H2O(s) for Zn and Cu3(PO4)2(s) and Cu2CO3(OH)2(s) for Cu (Figure 9).
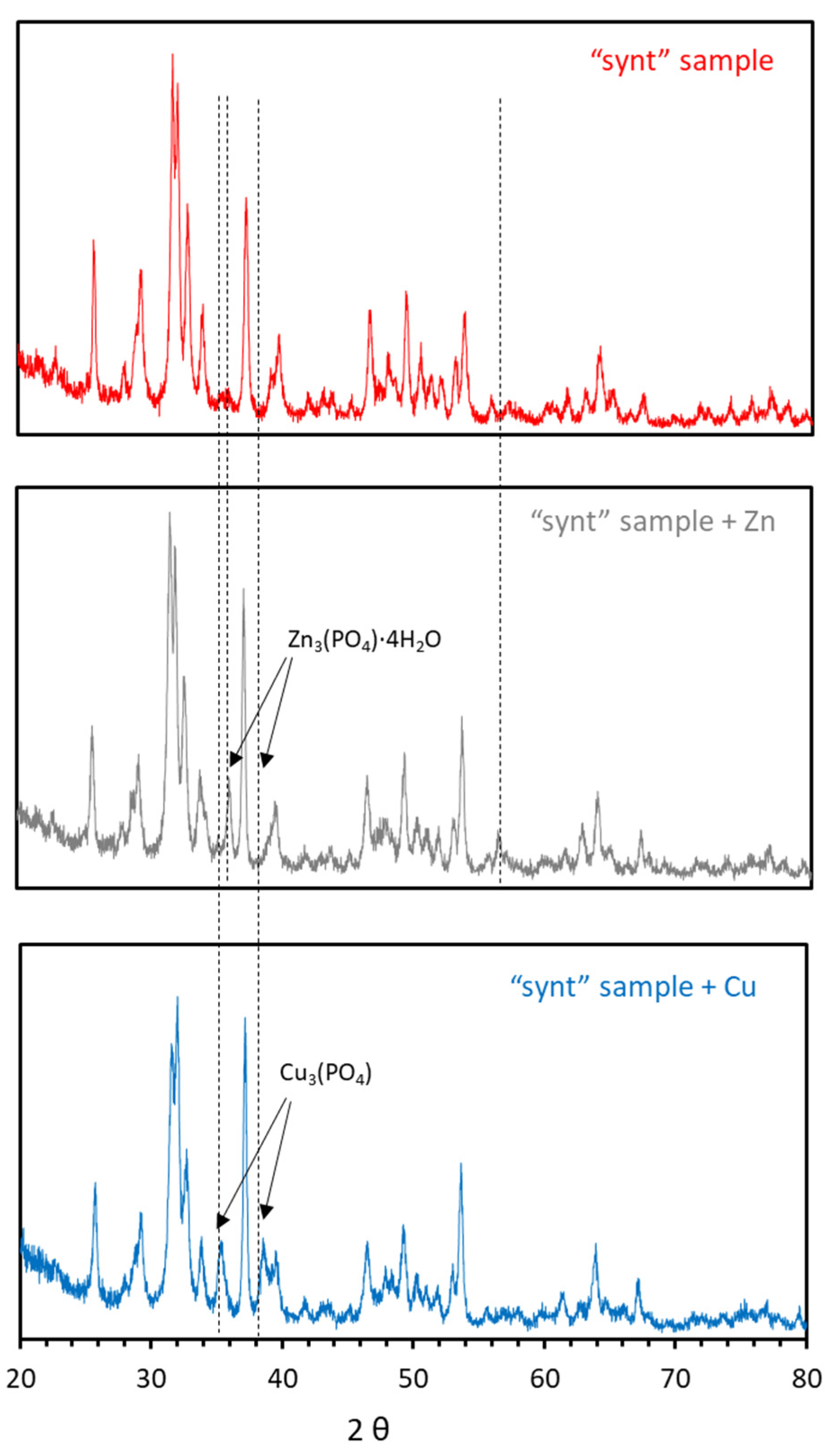
XRD analysis performed on HAP loaded with Zn and Cu from the sorption experiments did not offer any peak attributable to new phases (not shown), probably due to their low crystallinity. However, XRD analysis of these samples after calcination at 700 °C for 3 h revealed the appearance of peaks associated to Zn3(PO4)2·4H2O (hopeite) for Zn, and to Cu3(PO4)2 for Cu (Figure 10). The appearance of these peaks confirmed the precipitation of Zn and Cu in the form of phosphates (it must be kept in mind that calcination might have altered the precipitated new phases, e.g., decarbonation by loss of CO2, and that the observed phases by XRD can be only a part of the precipitated phases). Hopeite formation in sorption experiments of Zn onto HAP has also been observed by Stötzel et al. [50] and Sheha [35]. Although not in the present study, hydrozincite has also been detected by Sheha [35] and Meski et al. [36]. With regard to Cu, Cu(OH)2 and Cu2(PO4)(OH) (libethenite) have been detected by Šljivić et al. [43].
Further experiments were performed to experimentally confirm the occurrence of Zn and Cu precipitation in the studied conditions. With the purpose of isolating the eventual precipitation mechanism, HAP amounts (0.1 g) were weighted and equilibrated with deionized water (50 mL) at pH 4.25 for 24 h. Afterwards, the generated lixiviates were filtrated, and then, in the absence of HAP (i.e., without the possibility of sorption occurrence), Zn (or Cu) was added to achieve a final metal concentration of 50 mg/L. pH was adjusted to achieve different final values of 2.2, 6.7 and 8.0. Under these conditions, any disappearance of metal from the solution could only be attributed to precipitation. The analyses showed that at pH 2.2, neither Zn nor Cu were barely removed (<5%), but Zn was removed by 33% at pH 6.7 and by 99% at pH 8.0, while Cu was removed by 97% at pH 6.7 and >99% at pH 8.0. These findings were in agreement with the Zn and Cu speciation diagrams (Figure 9), except for Zn at pH 6.7, for which the observed removal was lower than the theoretical one.
4. Conclusions
Calcite MPs were efficiently coated with a HAP layer following a synthesis route based on two successive precipitations of HAP, with the first involving ethanol 0.5%. Since bare areas of calcite occurred, at pH 4.6 (that of the water to be treated), calcite partially dissolved, raising pH to values >9. The synthesized material showed high capacity in sorbing Zn and Cu from acidic solutions, making it potentially attractive for the removal of divalent heavy metals from contaminated water. The sorption isotherm was well fitted by the Langmuir model, with qmax of 34.97 mg/g for Zn and 60.24 mg/g for Cu, indicating more affinity of HAP towards Cu than towards Zn. Sorption of Zn and Cu onto the synthesized HAP followed a pseudo-second order kinetics, with k2 of 0.014 g/(mg·min) for Zn and 0.057 g/(mg·min) for Cu. On the other hand, sonication of the synthesized material seemed to lead to some breakage of agglomerated particles (decrease of mean diameter agglomerate from 36 to 23 μm), which resulted in a slight increase of qmax for Zn (37.88 g/mg) without affecting that of Cu. The mechanisms behind the sorption of Zn and Cu onto HAP were diverse and mainly included surface complexation, ion exchange and precipitation of new Zn- and Cu-containing phases. These mechanisms acted together, and it was difficult to quantitatively estimate the contribution of each of them to the overall removal of Zn and Cu.
In view of this performance, we anticipate that the HAP-coated calcite MPs prepared in this study have the potential to remediate heavy metal-contaminated water in ex-situ and, more promisingly, in-situ treatments. When injected into the subsoil for in-situ treatment, these HAP-coated calcite MPs may avoid complications that other MPs and NPs aiming at the removal of heavy metals face, as reported by the relatively abundant fundamental research but also by the currently scarce experience in field applications. In fact, NPs based on carbonaceous materials do not always exhibit high metal sorption capacities unless they are functionalized, while zero-valent iron NPs have a strong tendency to aggregate, which hinders their migration through the contaminated plume, and to have their surface passivated by precipitated of Fe-(oxy)hydroxide, which limits the reactivity of zero-valent iron. Despite this anticipated advantage of HAP-based particles, the limited availability of reports on their application for the removal of heavy metals from water makes further studies necessary, both at laboratory and field scales, before drawing major conclusions on the remediation constraints and environmental implications of their application.
On the other hand, it is acknowledged that one problem of NPs is the relatively high cost of synthesizing them. In this sense, and as an innovative aspect of the study, the synthesis route of HAP (based on solid calcite contacted with a phosphate solution instead of the conventional and widely employed wet chemical precipitation, which is based on the use of soluble Ca2+-salts) can allow to save huge amounts of soluble Ca-chemicals and make the process more cost-efficient and improve its competitiveness.
Author Contributions
Conceptualization: O.G., V.M., C.V. and M.M.M.; Methodology: O.G. and V.M.; Formal analysis: V.M. and R.M.D.; Investigation: V.M. and M.M.M.; Resources: O.G. and V.M.; Data curation: O.G., V.M., C.V., M.M.M. and R.M.D.; Writing—Original draft preparation: O.G.; Writing—Review and editing: V.M., C.V. and J.O.M.; Supervision: V.M. and J.O.M.; Funding acquisition: V.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Spanish Ministry of Science, Innovation and Universities and European Funds FEDER (AEI/FEDER) through the project NANOREMOV (CGL2017-87216-C4-3-R). Financial support was also received from the Agency for University Grants Administration and Research (AGAUR) of the Catalan Government through the Research Groups Support program (2014-SGR-050).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study is available on request from the corresponding author.
Acknowledgments
The authors thank A. Marín, R. Ramos and A. Guillaumet for their laboratory assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Burri, N.M.; Weatherl, R.; Moeck, C.; Schirmer, M. A review of threats to groundwater quality in the Anthropocene. Sci. Total Environ. 2019, 684, 136–154. [Google Scholar] [CrossRef]
- Zhou, P.; Adeel, M.; Shakoor, N.; Guo, M.; Hao, Y.; Azeem, I.; Li, M.; Liu, M.; Rui, Y. Application of nanoparticles alleviates heavy metals stress and promotes plant growth: An overview. Nanomaterials 2021, 11, 26. [Google Scholar] [CrossRef]
- Vareda, J.P.; Valente, A.J.M.; Durães, L. Assessment of heavy metal pollution from anthropogenic activities and remediation strategies: A review. J. Environ. Manag. 2019, 246, 101–118. [Google Scholar] [CrossRef]
- Vidu, R.; Matei, E.; Predescu, A.M.; Alhalaili, B.; Pantilimon, C.; Tarcea, C.; Predescu, C. Removal of heavy metals from wastewaters: A challenge from current treatment methods to nanotechnology applications. Toxics 2020, 8, 101. [Google Scholar] [CrossRef]
- Carolin, C.F.; Kumar, P.S.; Saravanan, A.; Joshiba, G.J.; Naushad, M. Efficient techniques for the removal of toxic heavy metals from aquatic environment: A review. J. Environ. Chem. Eng. 2017, 5, 2782–2799. [Google Scholar] [CrossRef]
- Abdullah, N.; Yusof, N.; Lau, W.J.; Jaafar, J.; Ismail, A.F. Recent trends of heavy metal removal from water/wastewater by membrane technologies. J. Ind. Eng. Chem. 2019, 76, 17–38. [Google Scholar] [CrossRef]
- Kaushik, A.; Singh, A. Metal removal and recovery using bioelectrochemical technology: The major determinants and opportunities for synchronic wastewater treatment and energy production. J. Environ. Manag. 2020, 270, 110826. [Google Scholar] [CrossRef] [PubMed]
- Malik, L.A.; Bashir, A.; Qureashi, A.; Pandith, A.H. Detection and removal of heavy metal ions: A review. Environ. Chem. Lett. 2019, 17, 1495–1521. [Google Scholar] [CrossRef]
- Waheed, A.; Baig, N.; Ullah, N.; Falath, W. Removal of hazardous dyes, toxic metal ions and organic pollutants from wastewater by using porous hyper-cross-linked polymeric materials: A review of recent advances. J. Environ. Manag. 2021, 287, 112360. [Google Scholar] [CrossRef] [PubMed]
- Adeleye, A.S.; Conway, J.R.; Garner, K.; Huang, Y.; Su, Y.; Keller, A.A. Engineered nanomaterials for water treatment and remediation: Costs, benefits, and applicability. Chem. Eng. J. 2016, 286, 640–662. [Google Scholar] [CrossRef] [Green Version]
- Henderson, A.D.; Demond, A.H. Long-term performance of zero-valent iron permeable reactive barriers: A critical review. Environ. Eng. Sci. 2007, 24, 401–423. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Fu, R.; Li, Q. Removal of inorganic contaminants in soil by electrokinetic remediation technologies: A review. J. Hazard. Mater. 2021, 401, 123345. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Tang, F.; Yan, J.; La, C.; Zheng, X.; Liu, W. Investigating the efficiency of microscale zero valent iron-based in situ reactive zone (mZVI-IRZ) for TCE removal in fresh and saline groundwater. Sci. Total Environ. 2018, 626, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Gibert, O.; Assal, A.; Devlin, H.; Elliot, T.; Kalin, R.M. Performance of a field-scale biological permeable reactive barrier for in-situ remediation of nitrate-contaminated groundwater. Sci. Total Environ. 2019, 659, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.S.; Shedbalkar, U.U.; Truskewycz, A.; Chopade, B.A.; Ball, A.S. Nanoparticles for environmental clean-up: A review of potential risks and emerging solutions. Environ. Technol. Innov. 2016, 5, 10–21. [Google Scholar] [CrossRef]
- Luo, J.; Yu, D.; Hristovski, K.D.; Fu, K.; Shen, Y.; Westerhoff, P.; Crittenden, J.C. Critical review of advances in engineering nanomaterial adsorbents for metal removal and recovery from water: Mechanism identification and engineering design. Environ. Sci. Technol. 2021, 55, 4287–4304. [Google Scholar] [CrossRef]
- Luo, J.; Fu, K.; Yu, D.; Hristovski, K.D.; Westerhoff, P.; Crittenden, J.C. Review of advances in engineering nanomaterial adsorbents for metal removal and recovery from water: Synthesis and microstructure impacts. ACS EST Eng. 2021, 1, 623–661. [Google Scholar] [CrossRef]
- Thanh, D.N.; Novák, P.; Vejpravova, J.; Vu, H.N.; Lederer, J.; Munshi, T. Removal of copper and nickel from water using nanocomposite of magnetic hydroxyapatite nanorods. J. Magn. Magn. Mater. 2018, 456, 451–460. [Google Scholar] [CrossRef]
- Cai, C.; Zhao, M.; Yu, Z.; Rong, H.; Zhang, C. Utilization of nanomaterials for in-situ remediation of heavy metal(loid) contaminated sediments: A review. Sci. Total Environ. 2019, 662, 205–217. [Google Scholar] [CrossRef]
- Ou, M.-Y.; Ting, Y.; Ch’ng, B.-L.; Chen, C.; Cheng, Y.-H.; Chang, T.-C.; Hsi, H.-C. Using mixed active capping to remediate multiple potential toxic metal contaminated sediment for reducing environmental risk. Water 2020, 12, 1886–1900. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Fihri, A.; Len, C.; Varma, R.S.; Solhy, A. Hydroxyapatite: A review of syntheses, structure and applications in heterogeneous catalysis. Coordin. Chem. Rev. 2017, 347, 48–76. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Shen, W.; Cui, J. Kinetics of hydroxyapatite precipitation at pH 10 to 11. Biomaterials 2001, 22, 301–3016. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.; Gong, H.; Jiang, X.; Wang, H.; Li, K. Effects of synthesis conditions on the morphology of hydroxyapatite nanoparticles produced by wet chemical process. Powder Technol. 2010, 203, 315–321. [Google Scholar] [CrossRef]
- Smičiklas, I.; Onjia, A.; Raičević, S.; Janaćković, Ð.; Mitrić, M. Factors influencing the removal of divalent cations by hydroxyapatite. J. Hazard. Mater. 2008, 152, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Marchegiani, F.; Cibej, E.; Vergni, P.; Tosi, G.; Fermani, S.; Falini, G. Hydroxyapatite synthesis from biogenic calcite single crystals into phosphate solutions at ambient conditions. J. Cryst. Growth 2009, 311, 4219–4225. [Google Scholar] [CrossRef]
- Ni, M.; Ratner, B.D. Nacre surface transformation to hydroxyapatite in a phosphate buffer solution. Biomaterials 2003, 24, 4323–4331. [Google Scholar] [CrossRef]
- Guo, Y.P.; Zhou, Y. Conversion of nacre powders to apatite in phosphate buffer solutions at low temperatures. Mater. Chem. Phys. 2007, 106, 88–94. [Google Scholar] [CrossRef]
- Pham Minh, D.; Lyczko, N.; Sebei, H.; Nzihou, A.; Sharrock, P. Synthesis of calcium hydroxyapatite from calcium carbonate and different orthophosphate sources: A comparative study. Mater. Sci. Eng. B 2012, 177, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Naidu, S.; Scherer, G.W. Nucleation, growth and evolution of calcium phosphate films on calcite. J. Colloid Interf. Sci. 2014, 435, 128–137. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y. Artificial hydroxyapatite film for the conservation of outdoor marble artworks. Mater. Lett. 2014, 124, 201–203. [Google Scholar] [CrossRef]
- Graziani, G.; Sassonia, E.; Franzonia, E.; Scherer, G.W. Hydroxyapatite coatings for marble protection: Optimization of calcite covering and acid resistance. Appl. Surf. Sci. 2016, 368, 241–257. [Google Scholar] [CrossRef]
- Ivanets, A.; Kitikova, N.; Shashkova, I.; Matrunchik, Y.; Kul’bitskaya, L.; Sillanpää, M. Non-acidic synthesis of phosphatized dolomite and its sorption behaviour towards Pb2+, Zn2+, Cu2+, Cd2+, Ni2+, Sr2+ and Co2+ ions in multicomponent aqueous solution. Environ. Technol. Innov. 2016, 6, 152–164. [Google Scholar] [CrossRef]
- Fiol, N.; Villaescusa, I. Determination of sorbent point zero charge: Usefulness in sorption studies. Environ. Chem. Lett. 2009, 7, 79–84. [Google Scholar] [CrossRef]
- Sheha, R.R. Sorption behavior of Zn(II) ions on synthesized hydroxyapatites. J. Colloid Interf. Sci. 2007, 310, 18–26. [Google Scholar] [CrossRef]
- Meski, S.; Ziani, S.; Khireddine, H.; Boudboub, S.; Zaidi, S. Factorial design analysis for sorption of zinc on hydroxyapatite. J. Hazard. Mater. 2011, 186, 1007–1017. [Google Scholar] [CrossRef]
- Klinkaewnarong, J.; Utara, S. Ultrasonic-assisted conversion of limestone into needle-like hydroxyapatite nanoparticles. Ultrason. Sonochem. 2018, 46, 18–25. [Google Scholar] [CrossRef]
- Lei, S.; Shi, Y.; Qiu, Y.; Che, L.; Xue, C. Performance and mechanisms of emerging animal-derived biochars for immobilization of heavy metals. Sci. Total Environ. 2019, 646, 1281–1289. [Google Scholar] [CrossRef]
- Ramasamy, V.; Anand, P.; Suresh, G. Synthesis and characterization of polymer-mediated CaCO3 nanoparticles using limestone: A novel approach. Adv. Powder Technol. 2018, 29, 818–834. [Google Scholar] [CrossRef]
- Salimi, M.N.; Bridson, R.H.; Grover, L.M.; Leeke, G.A. Effect of processing conditions on the formation of hydroxyapatite nanoparticles. Powder Technol. 2012, 218, 109–118. [Google Scholar] [CrossRef]
- Harding, I.S.; Rashid, N.; Hing, K.A. Surface charge and the effect of excess calcium ions on the hydroxyapatite surface. Biomaterials 2005, 26, 6818–6826. [Google Scholar] [CrossRef]
- Corami, A.; Mignardi, S.; Ferrini, V. Copper and zinc decontamination from single- and binary-metal solutions using hydroxyapatite. J. Hazard. Mater. 2007, 146, 164–170. [Google Scholar] [CrossRef]
- Šljivić, M.; Smičiklas, I.; Plećaš, I.; Mitrić, M. The influence of equilibration conditions and hydroxyapatite physico-chemical properties onto retention of Cu2+ ions. Chem. Eng. J. 2009, 148, 80–88. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, J.H.; Cui, Y.X.; Wang, S.Q.; Zhou, D.M. Effects of low-molecular-weight organic acids on Cu(II) adsorption onto hydroxyapatite nanoparticles. J. Hazard. Mater. 2009, 162, 1135–1140. [Google Scholar] [CrossRef]
- Feng, Y.; Gong, J.L.; Zeng, G.M.; Niu, Q.Y.; Zhang, H.Y.; Niu, C.G.; Deng, J.H.; Yan, M. Adsorption of Cd (II) and Zn (II) from aqueous solutions using magnetic hydroxyapatite nanoparticles as adsorbents. Chem. Eng. J. 2010, 162, 487–494. [Google Scholar] [CrossRef]
- Xu, Y.; Schwartz, F.W.; Traína, S.J. Sorption of Zn2+ and Cd2+ on hydroxyapatite surfaces. Environ. Sci. Technol. 1994, 28, 1472–1480. [Google Scholar] [CrossRef]
- Zhu, R.; Yu, R.; Yao, J.; Mao, D.; Xing, C.; Wang, D. Removal of Cd2+ from aqueous solutions by hydroxyapatite. Catal. Today 2008, 139, 94–99. [Google Scholar] [CrossRef]
- Smičiklas, I.D.; Milonjić, S.K.; Pfendt, P.; Raičević, S. The point of zero charge and sorption of cadmium (II) and strontium (II) ions on synthetic hydroxyapatite. Sep. Purif. Technol. 2000, 18, 185–194. [Google Scholar] [CrossRef]
- Fulmer, M.T.; Ison, I.C.; Hankermayer, C.R.; Constantz, B.R.; Ross, J. Measurements of the solubilities and dissolution rates of several hydroxyapatites. Biomaterials 2002, 23, 751–755. [Google Scholar] [CrossRef]
- Stötzel, C.; Müller, F.A.; Reinert, F.; Niederdraenk, F.; Barralet, J.E.; Gbureck, U. Ion adsorption behaviour of hydroxyapatite with different crystallinities. Colloid Surface B 2009, 74, 91–95. [Google Scholar] [CrossRef]
- Chemical Equilibrium Software Hydra and Medusa. Inorganic Chemistry Department, Royal Institute of Technology: Stockholm, Sweden. Available online: https://www.kth.se/che/medusa (accessed on 3 February 2021).
Figure 1.
FTIR spectra (a) and the XRD spectra (b) of the selected synthesized material and, for comparison, calcite and a commercial HAP (mHAPcom).
Figure 1.
FTIR spectra (a) and the XRD spectra (b) of the selected synthesized material and, for comparison, calcite and a commercial HAP (mHAPcom).

Figure 2.
SEM images of the “synt” sample (a,b), “son” sample (c), mHAPcom (d) and nHAPcom (e).

Figure 3.
Volumetric particle size distributions (PSD) of samples “synt”, “son”, mHAPcom and nHAPcom.
Figure 3.
Volumetric particle size distributions (PSD) of samples “synt”, “son”, mHAPcom and nHAPcom.

Figure 4.
Experimental immersion technique curves corresponding to “synt” and mHAPcom samples for the determination of pHPZC.
Figure 4.
Experimental immersion technique curves corresponding to “synt” and mHAPcom samples for the determination of pHPZC.

Figure 5.
Sorption of Zn and Cu onto the synthesized material (“synt” sample) as a function of time. Symbols represent experimental data and fitting lines were plotted using the pseudo-second order kinetic model.
Figure 5.
Sorption of Zn and Cu onto the synthesized material (“synt” sample) as a function of time. Symbols represent experimental data and fitting lines were plotted using the pseudo-second order kinetic model.

Figure 6.
Sorption isotherms for Zn (a) and Cu (b) onto the sorbents evaluated in this study. Symbols represent experimental data and fitting lines are the isotherms derived from the Langmuir equations.
Figure 6.
Sorption isotherms for Zn (a) and Cu (b) onto the sorbents evaluated in this study. Symbols represent experimental data and fitting lines are the isotherms derived from the Langmuir equations.

Figure 7.
Variations in pH and Ca2+ and PO43− concentrations following Zn sorption (a,b, respectively) and Cu (c,d, respectively) sorption.
Figure 7.
Variations in pH and Ca2+ and PO43− concentrations following Zn sorption (a,b, respectively) and Cu (c,d, respectively) sorption.

Figure 8.
Relationship between the number of moles of released Ca2+ vs the number of moles of adsorbed Zn (a) and Cu (b).
Figure 8.
Relationship between the number of moles of released Ca2+ vs the number of moles of adsorbed Zn (a) and Cu (b).

Figure 9.
Species distribution diagram as a function of pH for (a) Zn and (b) Cu with a total metal concentration of 50 mg/L in a matrix representative of the supernatants containing the counter-ion in the metal salt (Cl− for Zn and SO42− for Cu), CO32− and PO43−. Minor species are not shown. The grey bars indicate the pH value of experiments. Distribution diagrams have been plotted using the software packages Hydra and Medusa [51].
Figure 9.
Species distribution diagram as a function of pH for (a) Zn and (b) Cu with a total metal concentration of 50 mg/L in a matrix representative of the supernatants containing the counter-ion in the metal salt (Cl− for Zn and SO42− for Cu), CO32− and PO43−. Minor species are not shown. The grey bars indicate the pH value of experiments. Distribution diagrams have been plotted using the software packages Hydra and Medusa [51].

Figure 10.
XRD patterns of the synthesized material after contact with Zn and Cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Measured values of SBET surface areas for all studied sorbents.
| Sorbent | SBET (m2/g) |
|---|---|
| Initial calcite | 0.87 ± 0.01 |
| Synt | 58.25 ± 0.08 |
| Son | 53.68 ± 0.16 |
| mHAPcom | 60.77 ± 0.20 |
| nHAPcom | 18.93 ± 0.11 |
Table 2.
Comparison of the pseudo-first and pseudo-second order kinetic models for the sorption of Zn and Cu onto the synthesized material (“synt” sample).
Table 2.
Comparison of the pseudo-first and pseudo-second order kinetic models for the sorption of Zn and Cu onto the synthesized material (“synt” sample).
| Metal | Pseudo-First Order Kinetic Model | Pseudo-Second Order Kinetic Model | ||||
|---|---|---|---|---|---|---|
| qe | k1 | R2 | qe | k2 | R2 | |
| (mg/g) | (1/min) | (mg/g) | g/(mg·min) | |||
| Zn | 31.38 | 0.099 | 0.840 | 31.37 | 0.014 | 0.998 |
| Cu | 44.08 | 0.079 | 0.966 | 44.05 | 0.057 | 0.998 |
R2: correlation factor obtained from the linearized from of the kinetic models.
Table 3.
Summary of obtained Langmuir adsorption parameters.
| Metal | Sorbent | qmax | KL | R2 | |
|---|---|---|---|---|---|
| (mg/g) | (mg/m2) | (L/mg) | |||
| Zn | synt | 34.97 | 0.600 | 0.91 | 0.9978 |
| son | 37.88 | 0.706 | 1.47 | 0.9984 | |
| mHAPcom | 29.67 | 0.489 | 0.68 | 0.9985 | |
| nHAPcom | 18.98 | 1.003 | 0.85 | 0.9967 | |
| Cu | synt | 60.24 | 1.034 | 15.09 | 0.9905 |
| son | 60.24 | 1.122 | 13.83 | 0.9955 | |
| mHAPcom | 38.61 | 0.635 | 2.98 | 0.9997 | |
| nHAPcom | 19.27 | 1.018 | 9.88 | 0.9977 | |
Table 4.
Comparison of Langmuir sorption parameters against previous studies with synthesized HAP through wet chemical precipitation.
Table 4.
Comparison of Langmuir sorption parameters against previous studies with synthesized HAP through wet chemical precipitation.
| HAP Synthesis and Characteristics | Adsorption | Ref | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Zn | Cu | ||||||||
| Precursors | pH | T | SBET (m2/g) | pH | qmax (mg/g) | qmax (mg/m2) | qmax (mg/g) | qmax (mg/m2) | |
| Ca(OH)2 + H3PO4 | n.r. | 20 | 67 | 5.0 | 37.53 | 0.560 | - | - | [25] |
| Ca(NO3)2 + (NH4)HPO4 (+Fe3O4/Fe2O3) | 11 | 90 | 142.5 | 5.0 (a) | 140.6 (b) | 0.99 | - | - | [45] |
| Ca(OH)2 + H3PO4 | n.r | 100 | 76.6 | 6.0 (a) | 102.04 | 1.332 | - | - | [35] |
| 5.5 (a) | 37.27 | 0.750 | - | - | |||||
| Ca(NO3)2 + (NH4)2HPO4 | 11 | n.r. | n.r. | 5.0 (a) | 10.75 | n.r. | - | - | [36] |
| commercial | 77 | 6.0 | 37.14 | 0.482 | - | - | [46] | ||
| commercial | 50 | 6.6 | 95.89 | 1.92 | 76.49 | 1.53 | [42] | ||
| CaCl2 + NH4H2PO4 + EtOH (+Fe3O4) | 11 | 20 | 101.2 | 5.0 (a) | - | - | 48.78 | 0.482 | [18] |
| Ca(OH)2 + H3PO4 | n.r. | n.r | 58 | 5.0 | - | - | 37.17 | 0.641 | [43] |
| Ca(NO3)2 + H3PO4 + NH4+-salt | 10 | 40 | 49.7 | 4.5 (a) | - | - | 29.23 | 0.588 | [44] |
| 5.5 (a) | - | - | 37.30 | 0.751 | |||||
| Calcite + (NH4)2PO4 (+EtOH) | 8 | 25 | 58.28 | 4.6 | 34.97 | 0.600 | 60.24 | 1.034 | This study |
| Calcite + (NH4)2PO4 (+EtOH) (+ sonication) | 53.68 | 4.6 | 37.88 | 0.706 | 60.24 | 1.122 | |||
| Commercial (mHAPcom) | 60.77 | 4.6 | 29.67 | 0.489 | 38.61 | 0.635 | |||
| Commercial (nHAPcom) | 18.93 | 4.6 | 18.98 | 1.003 | 19.27 | 0.985 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gibert, O.; Valderrama, C.; Martínez, M.M.; Darbra, R.M.; Moncunill, J.O.; Martí, V. Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water. Water 2021, 13, 1493. https://doi.org/10.3390/w13111493
AMA Style
Gibert O, Valderrama C, Martínez MM, Darbra RM, Moncunill JO, Martí V. Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water. Water. 2021; 13(11):1493. https://doi.org/10.3390/w13111493
Chicago/Turabian StyleGibert, Oriol, César Valderrama, María M. Martínez, Rosa Mari Darbra, Josep Oliva Moncunill, and Vicenç Martí. 2021. "Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water" Water 13, no. 11: 1493. https://doi.org/10.3390/w13111493
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.