
Metabarcoding under Brine: Microbial Ecology of Five Hypersaline Lakes at Rottnest Island (WA, Australia)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
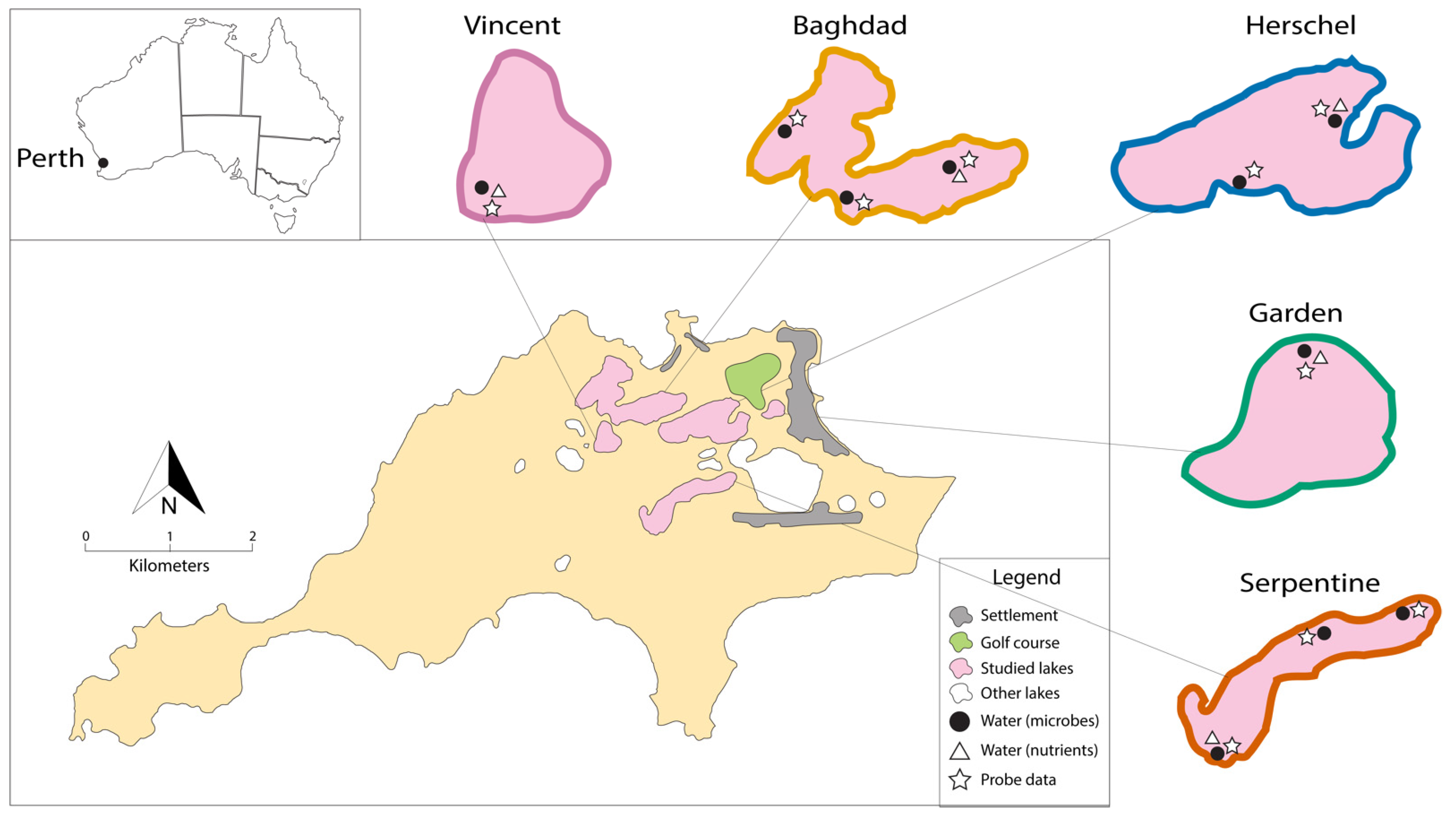
2.1. Study Area and Fieldwork
2.2. Laboratory Work, Data Analysis and Statistical Tests
2.2.1. Hydrochemical Analysis
2.2.2. Genetic Investigations
2.2.3. Statistical Analysis
3. Results
3.1. Biogeochemical Conditions
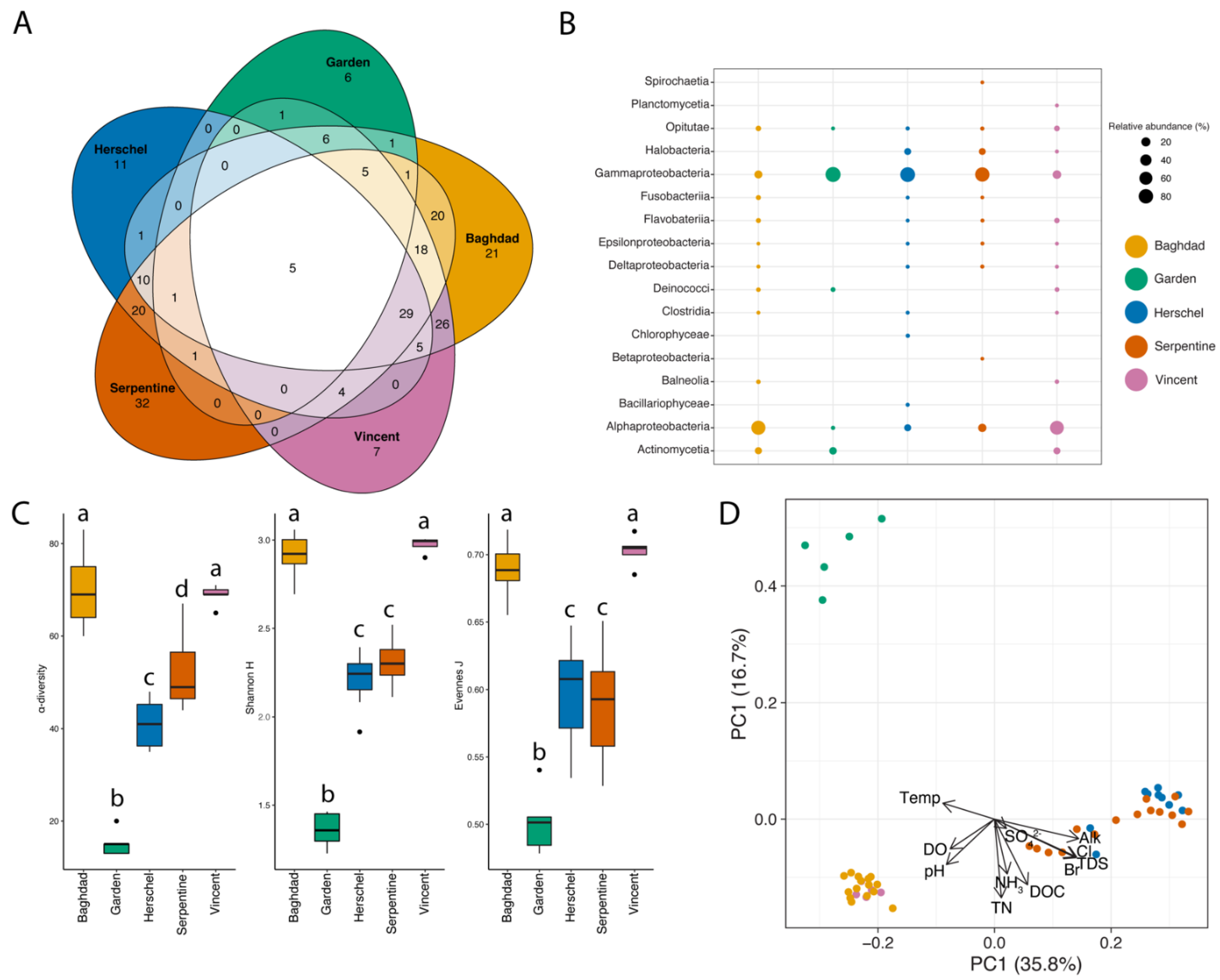
3.2. Microbial Community in Water
3.2.1. Community Assemblages and Environmental Drivers
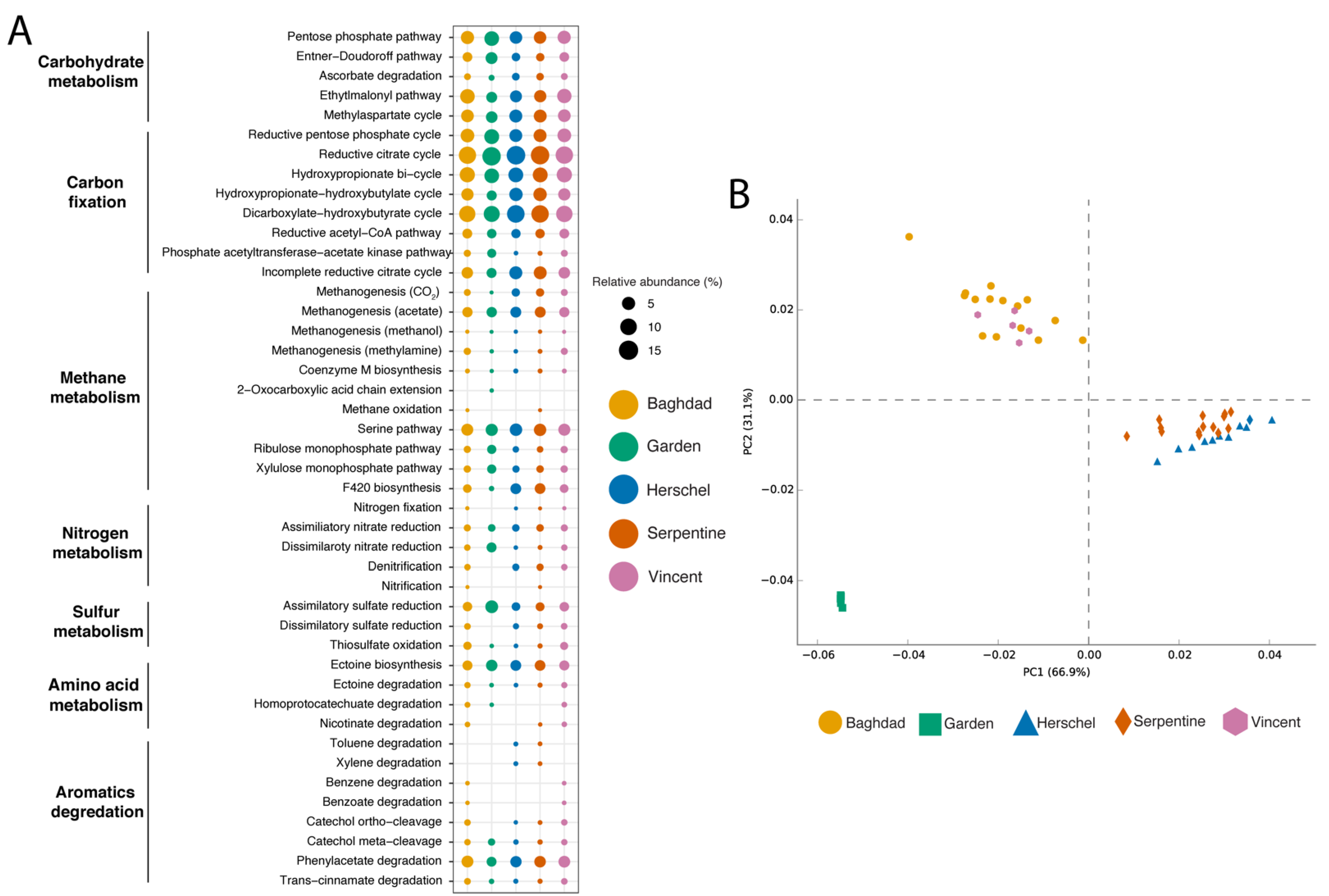
3.2.2. Predicted Functional Metabolisms
4. Discussion
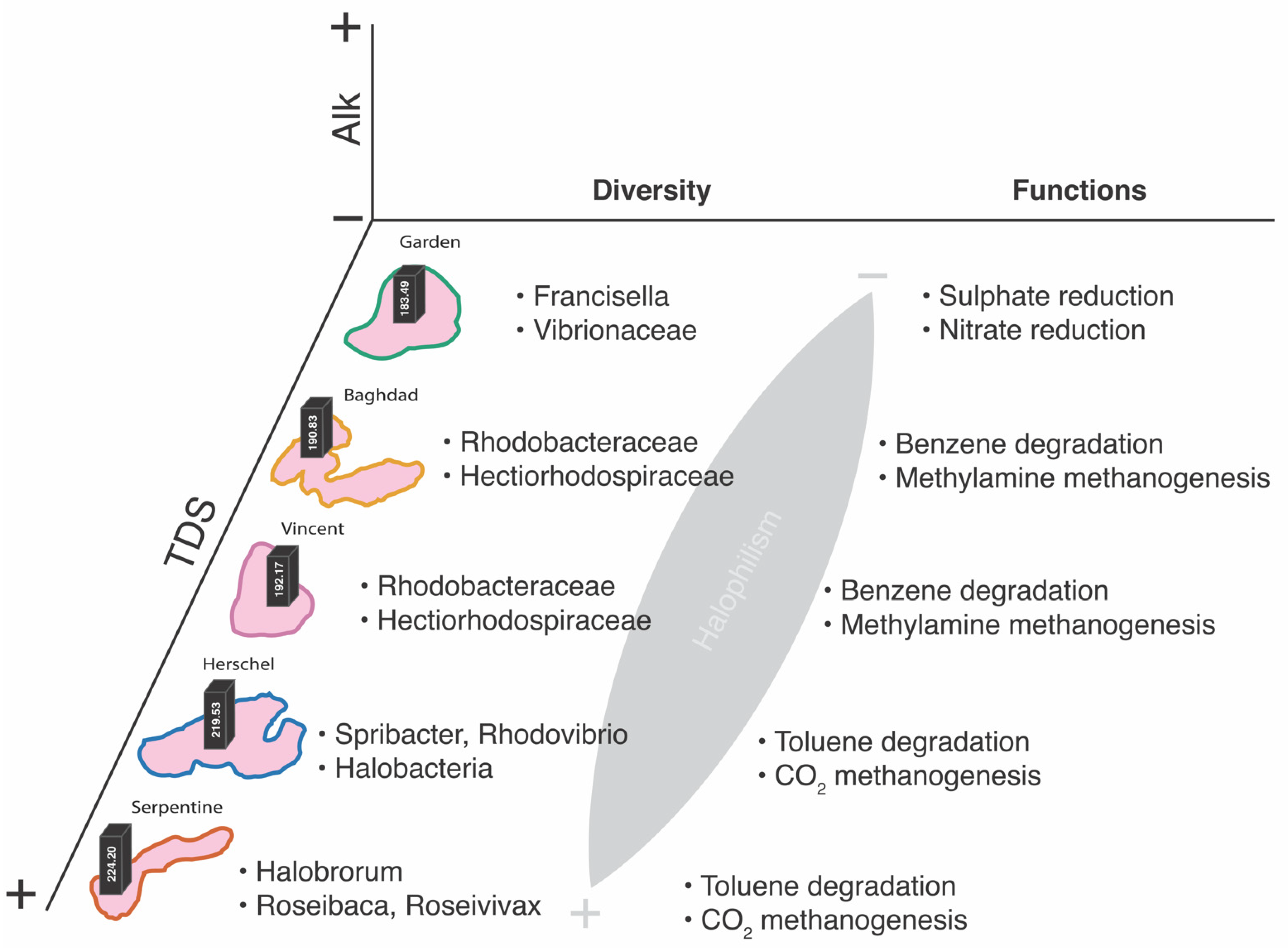
4.1. Biogeochemical Trends under High Salt
4.2. DNA Metabarcoding for Studying Hypersaline Microbial Communities
4.2.1. Taxonomic Patterns
4.2.2. Putative Metabolic Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Oren, A. Life in hypersaline environments. In Their World: A Diversity of Microbial Environments; Hurst, C.J., Ed.; Springer: Cham, Switzerland, 2016; Volume 1, pp. 301–339. [Google Scholar]
- Davila, A.F.; Duport, L.G.; Melchiorri, R.; Jaenchen, J.; Valea, S.; de Los Rios, A.; Fairen, A.G.; Moehlmann, D.; McKay, C.P.; Ascaso, C.; et al. Hygroscopic salts and the potential for life on Mars. Astrobiology 2010, 10, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Saccò, M.; White, N.E.; Harrod, C.; Salazar, G.; Aguilar, P.; Cubillos, C.F.; Meredith, K.; Baxter, B.K.; Oren, A.; Anufriieva, E.; et al. Salt to conserve: A review on the ecology and preservation of hypersaline ecosystems. Biol. Rev. 2021, in press. [Google Scholar]
- Saccò, M.; Blyth, A.J.; Humphreys, W.F.; Middleton, J.A.; White, N.E.; Campbell, M.; Mousavi-Derazmahalleh, M.; Laini, A.; Hua, Q.; Meredith, K.; et al. Tracking down carbon inputs underground from an arid zone Australian calcrete. PLoS ONE 2020, 15, e0237730. [Google Scholar] [CrossRef]
- Saccò, M.; Blyth, A.J.; Humphreys, W.F.; Cooper, S.J.; White, N.E.; Campbell, M.; Mousavi-Derazmahalleh, M.; Hua, Q.; Mazumder, D.; Smith, C.; et al. Rainfall as a trigger of ecological cascade effects in an Australian groundwater ecosystem. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Deiner, K.; Lopez, J.; Bourne, S.; Holman, L.E.; Seymour, M.; Grey, E.K.; Lacoursière-Roussel, A.; Li, Y.; Renshaw, M.A.; Pfrender, M.E.; et al. Optimising the detection of marine taxonomic richness using environmental DNA metabarcoding: The effects of filter material, pore size and extraction method. Metabarcoding Metagenomics 2018, 2, e28963. [Google Scholar] [CrossRef]
- Holman, L.E.; de Bruyn, M.; Creer, S.; Carvalho, G.; Robidart, J.; Rius, M. Detection of introduced and resident marine species using environmental DNA metabarcoding of sediment and water. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.M.; Kitson, J.J.; Lunt, D.H.; Straw, N.A.; Pocock, M.J. Merging DNA metabarcoding and ecological network analysis to understand and build resilient terrestrial ecosystems. Funct. Ecol. 2016, 30, 1904–1916. [Google Scholar] [CrossRef] [Green Version]
- Van der Heyde, M.; Bunce, M.; Dixon, K.; Wardell-Johnson, G.; White, N.E.; Nevill, P. Changes in soil microbial communities in post mine ecological restoration: Implications for monitoring using high throughput DNA sequencing. Sci. Total Environ. 2020, 749, 142262. [Google Scholar] [CrossRef] [PubMed]
- Elbrecht, V.; Steinke, D. Scaling up DNA metabarcoding for freshwater macrozoobenthos monitoring. Freshw. Biol. 2019, 64, 380–387. [Google Scholar]
- Yang, J.; Zhang, X.; Xie, Y.; Song, C.; Zhang, Y.; Yu, H.; Burton, G.A. Zooplankton community profiling in a eutrophic freshwater ecosystem-lake tai basin by DNA metabarcoding. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Keck, F.; Vasselon, V.; Rimet, F.; Bouchez, A.; Kahlert, M. Boosting DNA metabarcoding for biomonitoring with phylogenetic estimation of operational taxonomic units’ ecological profiles. Mol. Ecol. Resour. 2018, 18, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.D. Salinity as a determinant of the structure of biological communities in salt lakes. Hydrobiologia 1998, 381, 191–201. [Google Scholar] [CrossRef]
- Telesh, I.; Schubert, H.; Skarlato, S. Life in the salinity gradient: Discovering mechanisms behind a new biodiversity pattern. Estuar. Coast. Shelf Sci. 2013, 135, 317–327. [Google Scholar] [CrossRef]
- Messager, M.L.; Lehner, B.; Grill, G.; Nedeva, I.; Schmitt, O. Estimating the volume and age of water stored in global lakes using a geo-statistical approach. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S. Ecology of Hypersaline Microorganisms; National Institute of Oceanography: Goa, India, 2004. [Google Scholar]
- Finlayson, C.; Milton, G.R.; Prentice, R.C. Wetland types and distribution. In The Wetland Book II: Distribution, Description and Conservation; Springer: Berlin/Heidelberg, Germany, 2018; pp. 19–35. [Google Scholar]
- Nichols, G.; Williams, E.; Paola, C. (Eds.) Sedimentary Processes, Environments and Basins: A Tribute to Peter Friend; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 22. [Google Scholar]
- Mann, K.H. Retrieved 25/2/2021. Saline Lakes. Encyclopedia Britannica. Available online: https://www.britannica.com/science/inland-water-ecosystem/Saline-lakes (accessed on 15 January 2021).
- Trick, J.K.; Stuart, M.; Reeder, S. Contaminated groundwater sampling and quality control of water analyses. In Environmental Geochemistry; Elsevier: Amsterdam, The Netherlands, 2008; pp. 29–57. [Google Scholar]
- Prasad, B.S.R.V.; Srinivasu, P.D.N.; Varma, P.S.; Raman, A.V.; Ray, S. Dynamics of dissolved oxygen in relation to saturation and health of an aquatic body: A case for Chilka Lagoon, India. J. Ecosyst. 2014. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, I.; Assouline, S.; Tanny, J.; Lensky, I.M.; Gertman, I.; Mor, Z.; Lensky, N.G. Seasonal and diurnal evaporation from a deep hypersaline lake: The Dead Sea as a case study. J. Hydrol. 2018, 562, 155–167. [Google Scholar] [CrossRef]
- Didari, M.; Bagheri, M.; Amoozegar, M.A.; Bouzari, S.; Babavalian, H.; Tebyanian, H.; Hassanshahian, M.; Ventosa, A. Diversity of halophilic and halotolerant bacteria in the largest seasonal hypersaline lake (Aran-Bidgol-Iran). J. Environ. Health Sci. Eng. 2020, 18, 961–971. [Google Scholar] [CrossRef]
- López-González, P.J.; Guerrero, F.; Castro, M.C. Seasonal fluctuations in the plankton community in a hypersaline temporary lake (Honda, Southern Spain). Int. J. Salt Lake Res. 1997, 6, 353–371. [Google Scholar] [CrossRef]
- Podell, S.; Emerson, J.B.; Jones, C.M.; Ugalde, J.A.; Welch, S.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. Seasonal fluctuations in ionic concentrations drive microbial succession in a hypersaline lake community. ISME J. 2014, 8, 979–990. [Google Scholar] [CrossRef]
- Micklin, P.P. Desiccation of the Aral Sea: A water management disaster in the Soviet Union. Science 1988, 241, 1170–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micklin, P. The past, present, and future Aral Sea. Lakes Reserv. Res. Manag. 2010, 15, 193–213. [Google Scholar] [CrossRef]
- Bryan, E.; Meredith, K.T.; Baker, A.; Post, V.E.; Andersen, M.S. Island groundwater resources, impacts of abstraction and a drying climate: Rottnest Island, Western Australia. J. Hydrol. 2016, 542, 704–718. [Google Scholar] [CrossRef]
- Mendes Monteiro, J.; Vogwill, R.; Bischoff, K.; Gleeson, D.B. Comparative metagenomics of microbial mats from hypersaline lakes at Rottnest Island (WA, Australia), advancing our understanding of the effect of mat community and functional genes on microbialite accretion. Limnol. Oceanogr. 2020, 65, 293–309. [Google Scholar] [CrossRef]
- Bischoff, K.; Sirantoine, E.; Wilson, M.E.; George, A.D.; Mendes Monteiro, J.; Saunders, M. Spherulitic microbialites from modern hypersaline lakes, Rottnest Island, Western Australia. Geobiology 2020, 18, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Playford, P.E.; Leech, R.E.J.; Kendrick, G.W. Geology and Hydrology of Rottnest Island; Geological Survey of Western Australia: Perth, Australia, 1977; p. 113. [Google Scholar]
- Mylroie, J.E.; Carew, J.L.; Moore, A.I. Blue holes: Definition and genesis. Carbonates Evaporites 1995, 10, 225–233. [Google Scholar] [CrossRef]
- Collins, L.B.; Zhu, Z.R.; Wyrwoll, K.H.; Hatcher, B.G.; Playford, P.E.; Chen, J.H.; Eisenhauer, A.; Wasserburg, G.J. Late Quaternary evolution of coral reefs on a cool-water carbonate margin: The Abrolhos Carbonate Platforms, southwest Australia. Mar. Geol. 1993, 110, 203–212. [Google Scholar] [CrossRef]
- Kis-Papo, T.; Grishkan, I.; Oren, A.; Wasser, S.P.; Nevo, E. Spatiotemporal diversity of filamentous fungi in the hypersaline Dead Sea. Mycol. Res. 2001, 105, 749–756. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.; Pryer, K.M.; Miao, V.P.; Palmer, J.D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999, 46, 327–338. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ashton, B.; Buxton, S.; Cheung, M.; Cooper, A.; Duran, C.; Field, M.; Heled, J.; Kearse, M.; Markowitz, S.; et al. Geneious v5.4. 2011. Available online: http://www.geneious.com/ (accessed on 18 April 2021).
- Mousavi-Derazmahalleh, M.; Stott, A.; Lines, R.; Peverley, G.; Nester, G.; Simpson, T.; Zawierta, M.; De La Pierre, M.; Bunce, M.; Christophersen, C.T. eDNAFlow, an automated, reproducible and scalable workflow for analysis of environmental DNA sequences exploiting Nextflow and Singularity. Mol. Ecol. Resour. 2021, 21, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol. Lett. 2020, 19, 3316–3332. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013; Volume 201. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package; R Package Version 2.4-0; R Core Team: Vienna, Austria, 2016. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Martinez Arbizu, P. pairwiseAdonis: Pairwise Multilevel Comparison Using Adonis; R Package Version 0.0.1; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Cadotte, M.W.; Davies, T.J. Phylogenies in Ecology: A Guide to Concepts and Methods; Princeton University Press: Princeton, NJ, USA, 2016. [Google Scholar]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [Green Version]
- Pearse, W.D.; Purvis, A.; Cavender-Bares, J.; Helmus, M.R. Metrics and models of community phylogenetics. In Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 451–464. [Google Scholar]
- Chao, A.; Chiu, C.-H.; Jost, L. Phylogenetic diversity measures and their decomposition: A framework based on Hill numbers. In Biodiversity Conservation and Phylogenetic Systematics; Topics in Biodiversity and Conservation; Pellens, R., Grandcolas, P., Eds.; Springer: Cham, Switzerland, 2016; Volume 14. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthik, L.; Kumar, G.; Keswani, T.; Bhattacharyya, A.; Chandar, S.S.; Rao, K.B. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS ONE 2014, 3, e90972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Hay, M.; Paton, J. Cyanobacteria in benthic microbial communities in coastal salt lakes in Western Australia. Algol. Stud. 2009, 130, 125. [Google Scholar] [CrossRef]
- Mostofa, K.M.; Liu, C.Q.; Mottaleb, M.A.; Wan, G.; Ogawa, H.; Vione, D.; Yoshioka, T.; Wu, F. Dissolved organic matter in natural waters. In Photobiogeochemistry of Organic Matter; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–137. [Google Scholar]
- Shapovalova, A.A.; Khijniak, T.V.; Tourova, T.P.; Muyzer, G.; Sorokin, D.Y. Heterotrophic denitrification at extremely high salt and pH by haloalkaliphilic Gammaproteobacteria from hypersaline soda lakes. Extremophiles 2008, 12, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, D.Y.; Kuenen, J.G. Chemolithotrophic haloalkaliphiles from soda lakes. FEMS Microbiol. Ecol. 2005, 52, 287–295. [Google Scholar] [CrossRef]
- Yadav, A.K.; Vardhan, S.; Kashyap, S.; Yandigeri, M.; Arora, D.K. Actinomycetes diversity among rRNA gene clones and cellular isolates from Sambhar salt lake, India. Sci. World J. 2013. [Google Scholar] [CrossRef] [Green Version]
- Jose, P.A.; Jebakumar, S.R.D. Unexplored hypersaline habitats are sources of novel actinomycetes. Front. Microbiol. 2014, 5, 242. [Google Scholar] [CrossRef]
- Gupta, R.S.; Naushad, S.; Baker, S. Phylogenomic analyses and molecular signatures for the class Halobacteria and its two major clades: A proposal for division of the class Halobacteria into an emended order Halobacteriales and two new orders, Haloferacales ord. nov. and Natrialbales ord. nov., containing the novel families Haloferacaceae fam. nov. and Natrialbaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 1050–1069. [Google Scholar]
- Gupta, R.S.; Naushad, S.; Fabros, R.; Adeolu, M. A phylogenomic reappraisal of family-level divisions within the class Halobacteria: Proposal to divide the order Halobacteriales into the families Halobacteriaceae, Haloarculaceae fam. nov., and Halococcaceae fam. nov., and the order Haloferacales into the families, Haloferacaceae and Halorubraceae fam nov. Antonie Van Leeuwenhoek 2016, 109, 565–587. [Google Scholar]
- Newton, K.; Jeffries, T.C.; Smith, R.J.; Seymour, J.R.; Seuront, L.; Mitchell, J.G. Taxonomic and metabolic shifts in the Coorong bacterial metagenome driven by salinity and external inputs. J. Oceanol. Limnol. 2018, 36, 2033–2049. [Google Scholar] [CrossRef]
- López-Hermoso, C.; Rafael, R.; Sánchez-Porro, C.; Ventosa, A. Salinivibrio kushneri sp. nov., a moderately halophilic bacterium isolated from salterns. Syst. Appl. Microbiol. 2018, 41, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Wohlfarth, A.; Severin, J.; Galinski, E.A. The spectrum of compatible solutes in heterotrophic halophilic eubacteria of the family Halomonadaceae. Microbiology 1990, 136, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Pechous, R.D.; McCarthy, T.R.; Zahrt, T.C. Working toward the future: Insights into Francisella tularensis pathogenesis and vaccine development. Microbiol. Mol. Biol. Rev. 2009, 73, 684–711. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, C.A.; Kesterson, K.E.; Duncan, D.D.; Eshoo, M.W.; Wolcott, M. Identification and characterization of Francisella species from natural warm springs in Utah, USA. Lett. Appl. Microbiol. 2012, 54, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Garrity, G.M.; Bell, J.A.; Lilburn, T.; Family, I. Rhodobacteraceae fam. nov. In Bergey’s Manual of Systematic Bacteriology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 2, p. 161. [Google Scholar]
- Almeida-Dalmet, S.; Sikaroodi, M.; Gillevet, P.M.; Litchfield, C.D.; Baxter, B.K. Temporal study of the microbial diversity of the North Arm of Great Salt Lake, Utah, US. Microorganisms 2015, 3, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.J.; Howarth, R.W.; Garritt, R.H. Sulfur and carbon isotopes as tracers of salt-marsh organic matter flow. Ecology 1986, 67, 865–874. [Google Scholar] [CrossRef]
- Overmann, J.; Beatty, J.T.; Hall, K.J. Purple sulfur bacteria control the growth of aerobic heterotrophic bacterioplankton in a meromictic salt lake. Appl. Environ. Microbiol. 1996, 62, 3251–3258. [Google Scholar] [CrossRef] [Green Version]
- Bunn, S.E.; Edward, D.H.D. Seasonal meromixis in three hypersaline lakes on Rottnest Island, Western Australia. Mar. Freshw. Res. 1984, 35, 261–265. [Google Scholar] [CrossRef]
- Cayetano, R.D.A.; Park, J.; Kim, G.B.; Jung, J.H.; Kim, S.H. Enhanced anaerobic digestion of waste-activated sludge via bioaugmentation strategy-Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt2) analysis through hydrolytic enzymes and possible linkage to system performance. Bioresour. Technol. 2021, 332, 125014. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef] [Green Version]
- McGenity, T.J. Methanogens and methanogenesis in hypersaline environments. In Handbook of Hydrocarbon and Lipid Microbiology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 665–680. [Google Scholar]
- Sela-Adler, M.; Ronen, Z.; Herut, B.; Antler, G.; Vigderovich, H.; Eckert, W.; Sivan, O. Co-existence of methanogenesis and sulfate reduction with common substrates in sulfate-rich estuarine sediments. Front. Microbiol. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Kelley, C.A.; Poole, J.A.; Tazaz, A.M.; Chanton, J.P.; Bebout, B.M. Substrate limitation for methanogenesis in hypersaline environments. Astrobiology 2012, 12, 89–97. [Google Scholar] [CrossRef]
- Teufel, R.; Mascaraque, V.; Ismail, W.; Voss, M.; Perera, J.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. USA 2010, 107, 14390–14395. [Google Scholar] [CrossRef] [Green Version]
- Fathepure, B.Z. Recent studies in microbial degradation of petroleum hydrocarbons in hypersaline environments. Front. Microbiol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, A. The bioenergetic basis for the decrease in metabolic diversity at increasing salt concentrations: Implications for the functioning of salt lake ecosystems. In Saline Lakes; Springer: Dordrecht, The Netherlands, 2001; pp. 61–72. [Google Scholar]
- Bauld, J. Benthic microbial communities of Australian saline lakes. In Limnology in Australia; Springer: Dordrecht, The Netherlands, 1986; pp. 95–111. [Google Scholar]
- McMaster, K.; Savage, A.; Finston, T.; Johnson, M.S.; Knott, B. The recent spread of Artemia parthenogenetica in Western Australia. Hydrobiologia 2007, 576, 39–48. [Google Scholar] [CrossRef]
- Geddes, M.C. Occurrence of the brine shrimp Artemia (Anostraca) in Australia. Crustaceana 1979, 36, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.H.D. Inland water of Rottnest Island. J. R. Soc. West. Aust. 1983, 66, 41–47. [Google Scholar]
- Mather, S. Summer migrants—The importance of Rottnest Island for trans-equatorial bird species. J. East Asian Australas. Flyway 2020, 73–74, 29–36. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lake | pH | DO | Temperature | Alkalinity | NH3 | TDS | Cl− | Br− | SO42− | DOC | TN |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (mg L−1) | (°C) | (mg CaCO3 L−1) | (mg L−1) | (mg L−1) | (g L−1) | (g L−1) | (g L−1) | (mg L−1) | (mg L−1) | ||
| Baghdad | 9.34 ± 0.26 a | 3.57 ± 0.85 a | 25.85 ± 0.57 a | 190.83 ± 4.45 a | 0.31 ± 0.01 a | 146.66 ± 4.32 a | 105.27 ± 0.16 a | 0.26 a | 7.62 ± 0.01 a | 50.95 ± 0.49 a | 7.85 ± 0.07 a |
| Garden | 6.70 ± 0.01 b | 2.93 ± 0.20 ab | 25.82 ± 0.04 a | 183.49 ± 7.56 a | 0.21 ± 0.01 bc | 118 ± 1.63 b | 81.22 ± 0.08 b | 0.2 c | 7.47 ± 0.01 b | 30.25 ± 0.35 b | 3.3 ± 0.14 b |
| Herschel | 6.49 ± 0.10 bd | 1.84 ± 0.03 b | 24.62 ± 0.14 a | 219.53 ± 7.12 b | 0.35 ± 0.04 a | 185.33 ± 3.40 c | 147.01 ± 0.24 c | 0.36 b | 4.03 ± 0.01 c | 63.6 ± 0.28 c | 8.25 ± 0.21 a |
| Serpentine | 6.41 ± 0.17 bd | 2.61 ± 0.92 ab | 21.08 ± 1.22 b | 224.20 ± 4.00 b | 0.27 ± 0.02 ac | 186.67 ± 0.94 c | 138.27 ± 0.26 d | 0.34 ± 0.01 b | 12.17 ± 0.02 d | 48.8 ± 0.28 d | 6.4 c |
| Vincent | 6.16 ± 0.05 cd | 3.06 ± 0.19 ab | 22.34 ± 0.10 b | 192.17 ± 4.00 a | 0.30 ± 0.02 a | 144 ± 1.63 a | 109.94 ± 2.29 a | 0.26 ± 0.01 a | 7.74 ± 0.01 e | 54.9 ± 0.71 e | 8.3 ± 0.14 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saccò, M.; White, N.E.; Campbell, M.; Allard, S.; Humphreys, W.F.; Pringle, P.; Sepanta, F.; Laini, A.; Allentoft, M.E. Metabarcoding under Brine: Microbial Ecology of Five Hypersaline Lakes at Rottnest Island (WA, Australia). Water 2021, 13, 1899. https://doi.org/10.3390/w13141899
Saccò M, White NE, Campbell M, Allard S, Humphreys WF, Pringle P, Sepanta F, Laini A, Allentoft ME. Metabarcoding under Brine: Microbial Ecology of Five Hypersaline Lakes at Rottnest Island (WA, Australia). Water. 2021; 13(14):1899. https://doi.org/10.3390/w13141899
Chicago/Turabian StyleSaccò, Mattia, Nicole E. White, Matthew Campbell, Sebastian Allard, William F. Humphreys, Paul Pringle, Farid Sepanta, Alex Laini, and Morten E. Allentoft. 2021. "Metabarcoding under Brine: Microbial Ecology of Five Hypersaline Lakes at Rottnest Island (WA, Australia)" Water 13, no. 14: 1899. https://doi.org/10.3390/w13141899