
Genome Sequencing of SARS-CoV-2 Allows Monitoring of Variants of Concern through Wastewater

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Wastewater Sample Collection and Processing
2.2. SARS-CoV-2 RNA Detection in Wastewater Samples
2.3. Amplicon Sequencing of Wastewater Samples
2.4. SARS-CoV-2 Genome Sequences from Luxembourg Patients
2.5. Lineage Specific Signature Mutations
2.6. Variant Calling
2.7. Haplotype Reconstruction
2.8. Statistical Analysis
3. Results
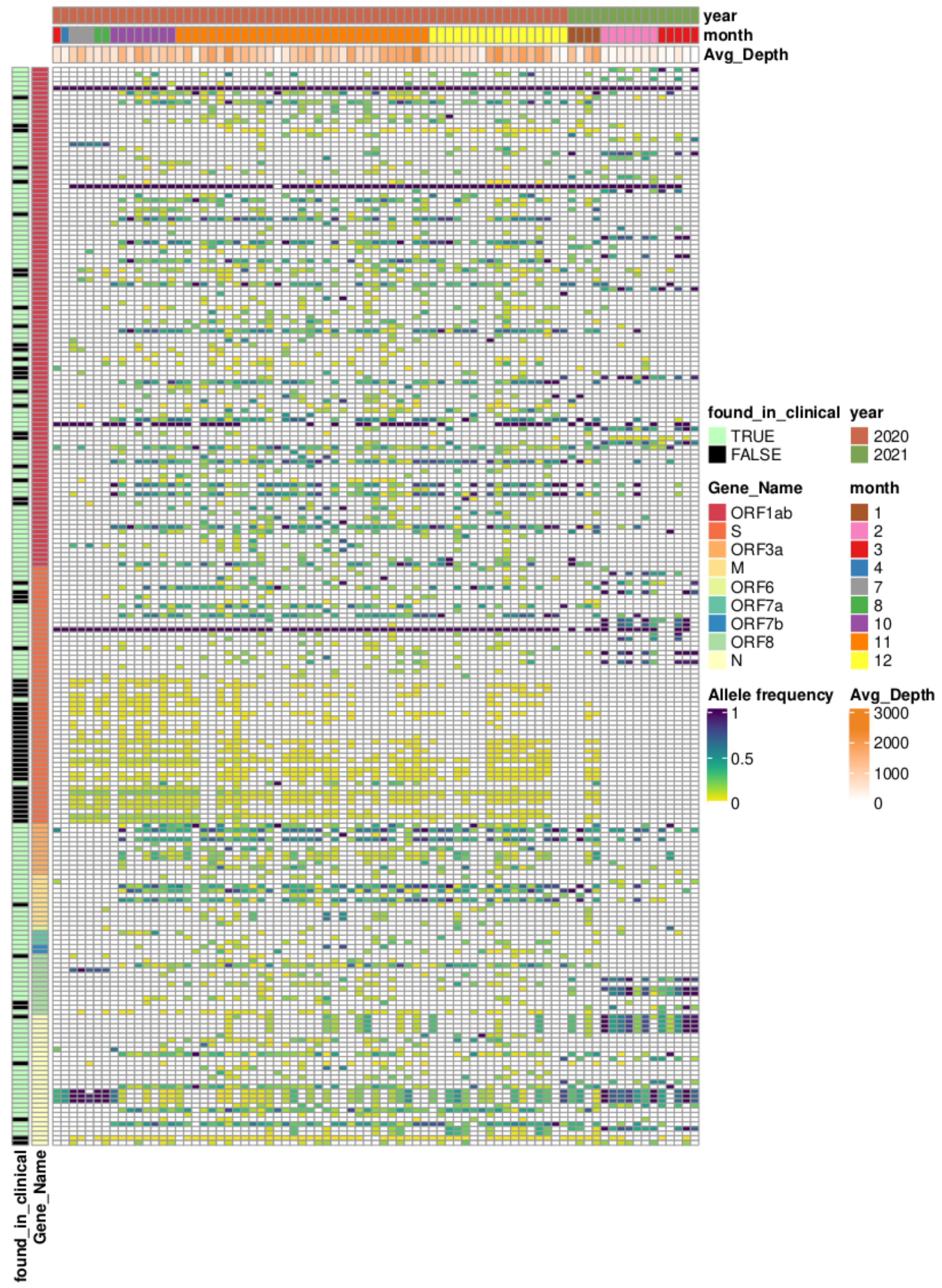
3.1. Overview of Available Clinical Sequences
3.2. Overview of Sequencing of Wastewater Samples
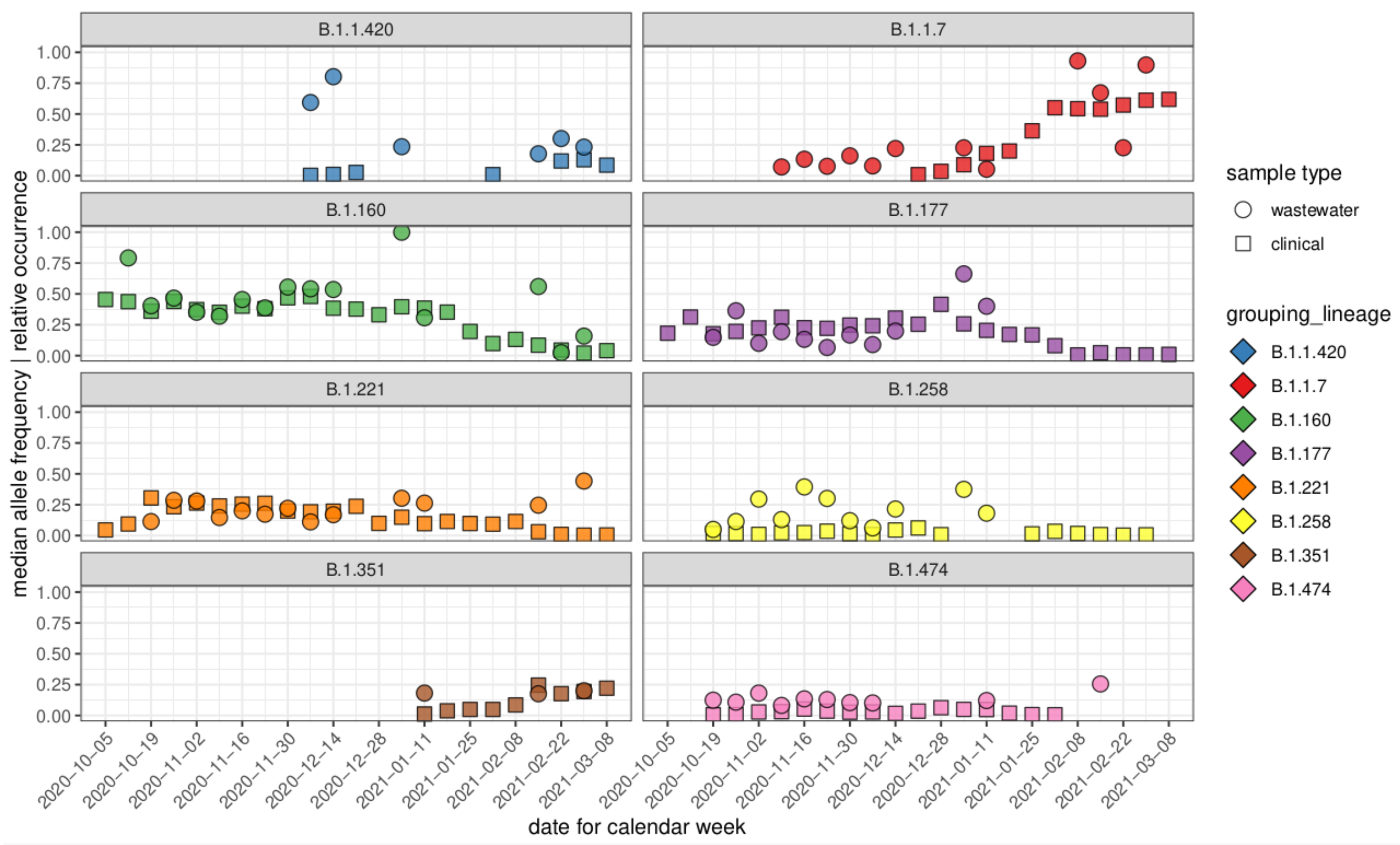
3.3. Comparison of Allele Frequencies and Relative Occurrence
3.4. Characteristic Mutations of VOCs
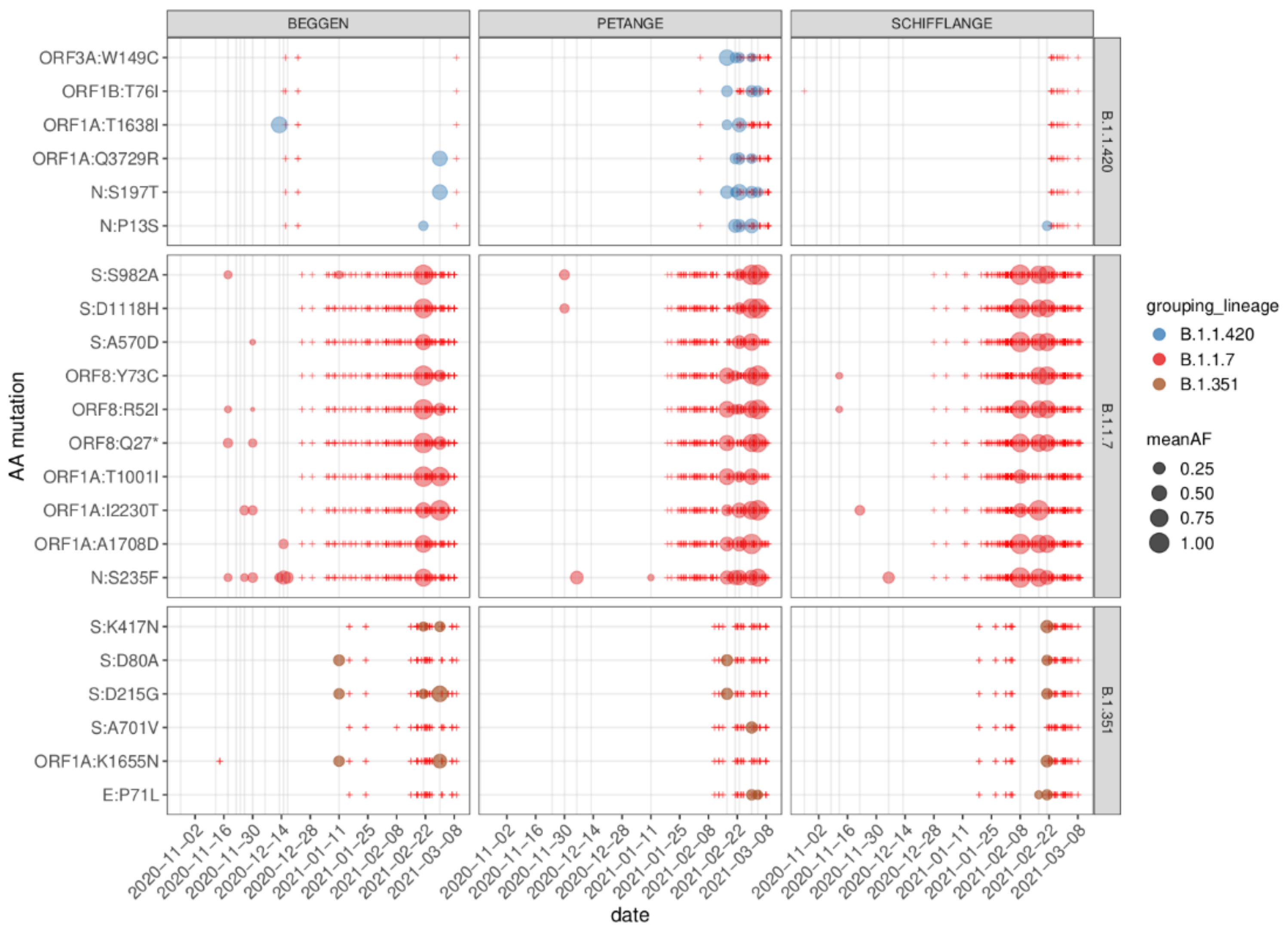
3.5. Regionally Specific Mutations
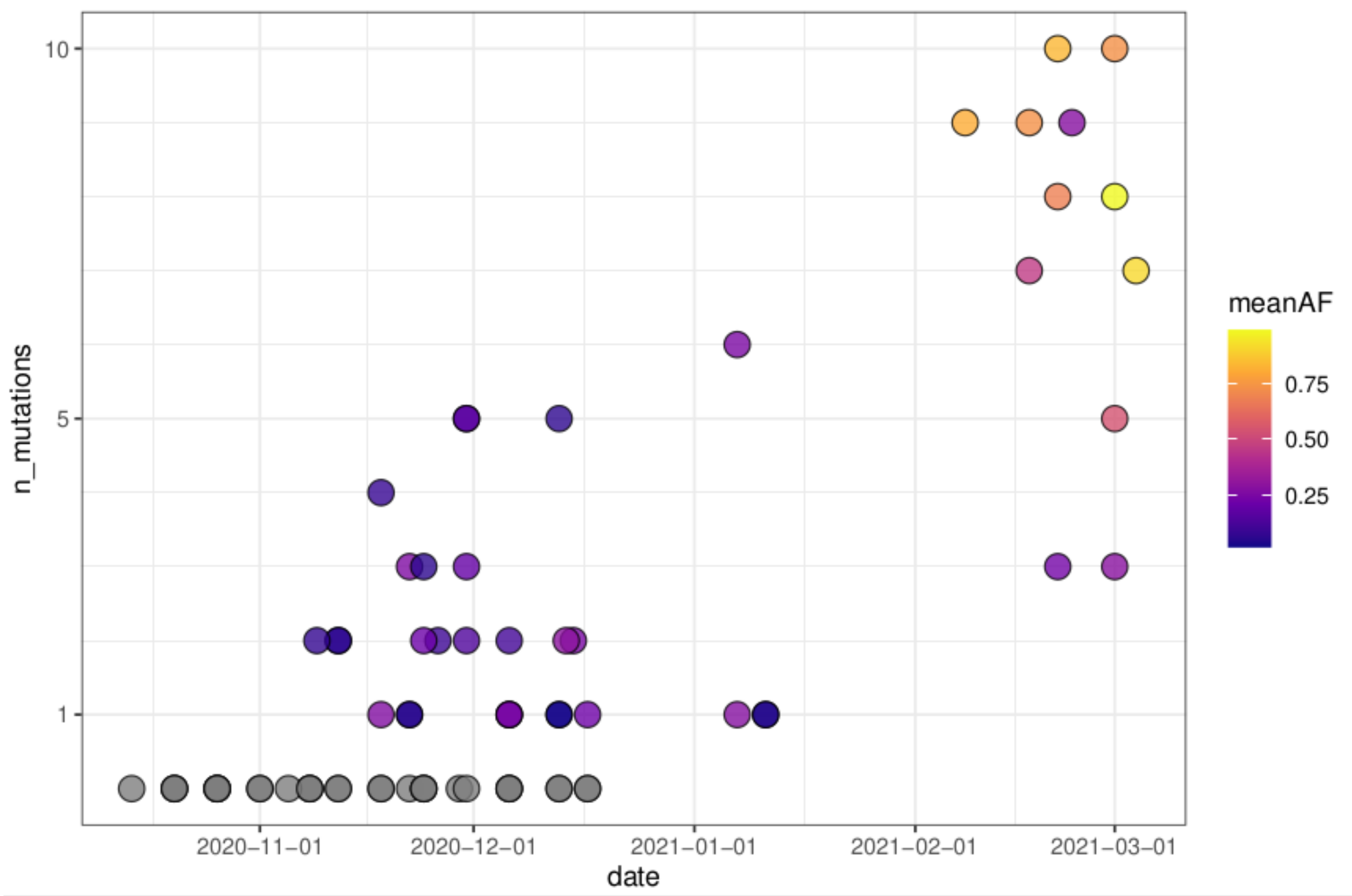
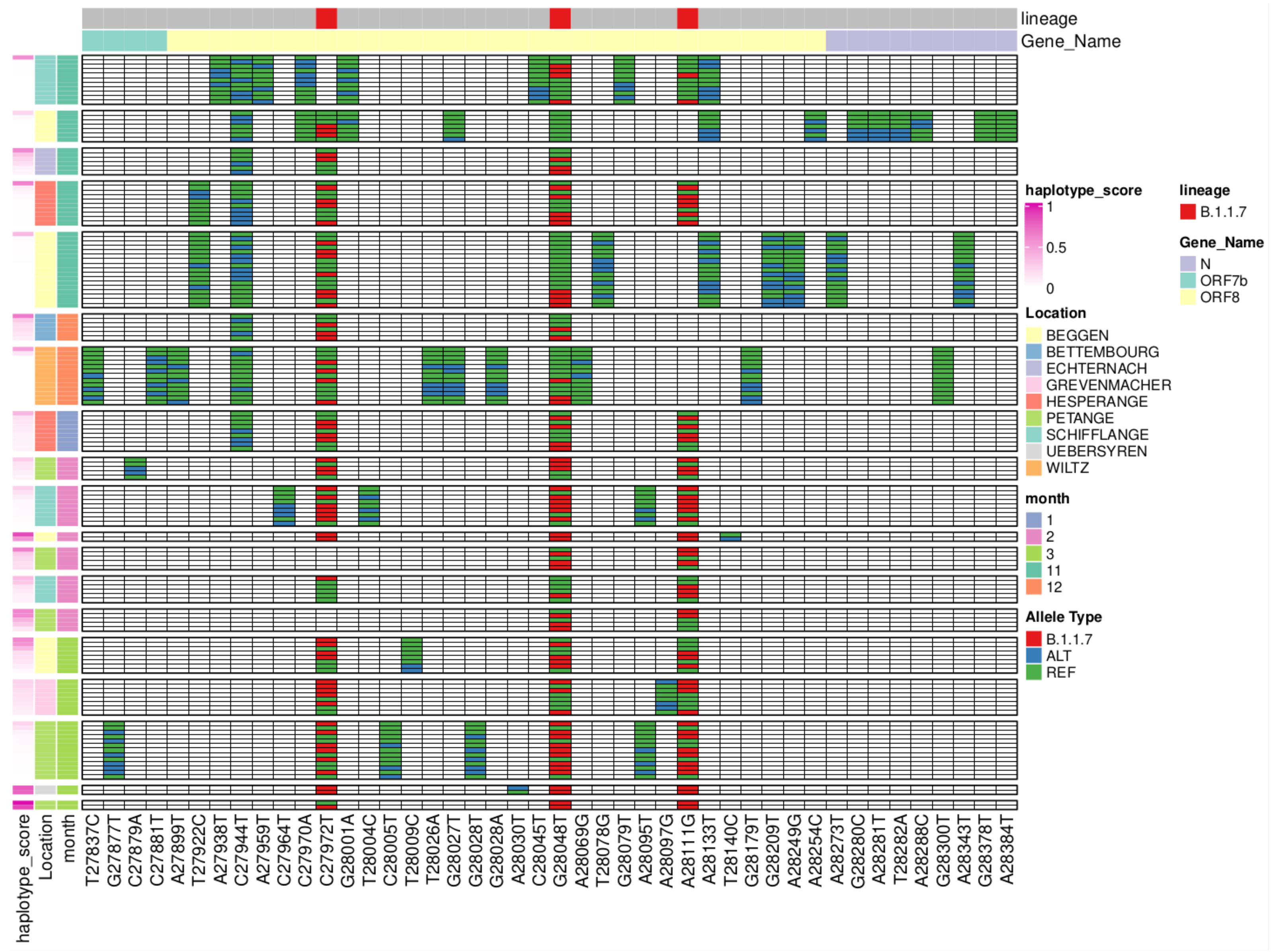
3.6. Reconstruction of Short Lineage-Specific Haplotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilmes, P.; Zimmer, J.; Schulz, J.; Glod, F.; Veiber, L.; Mombaerts, L.; Rodrigues, B.; Aalto, A.; Pastore, J.; Snoeck, C.J.; et al. SARS-CoV-2 transmission risk from asymptomatic carriers: Results from a mass screening programme in Luxembourg. Lancet Reg. Health Eur. 2021, 4, 100056. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Mao, Y.; Jones, R.M.; Tan, Q.; Ji, J.S.; Li, N.; Shen, J.; Lv, Y.; Pan, L.; Ding, P.; et al. Aerosol transmission of SARS-CoV-2? Evidence, prevention and control. Environ. Int. 2020, 144, 106039. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Parker, J.; Smits, S.; Underwood, J.; Dolwani, S. Persistent viral shedding of SARS-CoV-2 in faeces—A rapid review. Colorectal. Dis. 2020, 22, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Kitajima, M.; Omori, R. Duration of SARS-CoV-2 viral shedding in faeces as a parameter for wastewater-based epidemiology: Re-analysis of patient data using a shedding dynamics model. Sci. Total Environ. 2021, 769, 144549. [Google Scholar] [CrossRef]
- Crank, K.; Chen, W.; Bivins, A.; Lowry, S.; Bibby, K. Contribution of SARS-CoV-2 RNA shedding routes to RNA loads in wastewater. Sci. Total Environ. 2022, 806, 150376. [Google Scholar] [CrossRef]
- Sala-Comorera, L.; Reynolds, L.J.; Martin, N.A.; O’Sullivan, J.J.; Meijer, W.G.; Fletcher, N.F. Decay of infectious SARS-CoV-2 and surrogates in aquatic environments. Water Res. 2021, 201, 117090. [Google Scholar] [CrossRef]
- Han, J.; He, S. Urban flooding events pose risks of virus spread during the novel coronavirus (COVID-19) pandemic. Sci. Total Environ. 2021, 755, 142491. [Google Scholar] [CrossRef]
- Wathore, R.; Gupta, A.; Bherwani, H.; Labhasetwar, N. Understanding air and water borne transmission and survival of coronavirus: Insights and way forward for SARS-CoV-2. Sci. Total Environ. 2020, 749, 141486. [Google Scholar] [CrossRef]
- Paul, D.; Kolar, P.; Hall, S.G. A review of the impact of environmental factors on the fate and transport of coronaviruses in aqueous environments. NPJ Clean Water 2021, 4, 7. [Google Scholar] [CrossRef]
- Wurtzer, S.; Marechal, V.; Mouchel, J.; Maday, Y.; Teyssou, R.; Richard, E.; Almayrac, J.; Moulin, L. Evaluation of lockdown impact on SARS-CoV-2 dynamics through viral genome quantification in Paris wastewaters. MedRxiv 2020. [Google Scholar] [CrossRef]
- Peccia, J.; Zulli, A.; Brackney, D.E.; Grubaugh, N.D.; Kaplan, E.H.; Casanovas-Massana, A.; Ko, A.I.; Malik, A.A.; Wang, D.; Wang, M.; et al. Measurement of SARS-CoV-2 RNA in wastewater tracks community infection dynamics. Nat. Biotechnol. 2020, 38, 1164–1167. [Google Scholar] [CrossRef]
- Hillary, L.S.; Farkas, K.; Maher, K.H.; Lucaci, A.; Thorpe, J.; Distaso, M.A.; Gaze, W.H.; Paterson, S.; Burke, T.; Connor, T.R.; et al. Monitoring SARS-CoV-2 in municipal wastewater to evaluate the success of lockdown measures for controlling COVID-19 in the UK. Water Res. 2021, 200, 117214. [Google Scholar] [CrossRef]
- Medema, G.; Been, F.; Heijnen, L.; Petterson, S. Implementation of environmental surveillance for SARS-CoV-2 virus to support public health decisions: Opportunities and challenges. Curr. Opin. Environ. Sci. Health 2020, 17, 49–71. [Google Scholar] [CrossRef]
- Medema, G.; Heijnen, L.; Elsinga, G.; Italiaander, R.; Brouwer, A. Presence of SARS-Coronavirus-2 RNA in Sewage and Correlation with Reported COVID-19 Prevalence in the Early Stage of the Epidemic in The Netherlands. Environ. Sci. Technol. Lett. 2020, 7, 511–516. [Google Scholar] [CrossRef]
- Ahmed, W.; Bibby, K.; D’Aoust, P.M.; Delatolla, R.; Gerba, C.P.; Haas, C.N.; Hamilton, K.A.; Hewitt, J.; Julian, T.R.; Kaya, D.; et al. Differentiating between the possibility and probability of SARS-CoV-2 transmission associated with wastewater: Empirical evidence is needed to substantiate risk. FEMS Microbes 2021, 2. [Google Scholar] [CrossRef]
- Hart, O.E.; Halden, R.U. Computational analysis of SARS-CoV-2/COVID-19 surveillance by wastewater-based epidemiology locally and globally: Feasibility, economy, opportunities and challenges. Sci. Total Environ. 2020, 730, 138875. [Google Scholar] [CrossRef]
- McClary-Gutierrez, J.; Mattioli, M.; Marcenac, P.; Silverman, A.; Boehm, A.; Bibby, K.; Balliet, M.; de los Reyes, F.; Gerrity, D.; Griffith, J.; et al. SARS-CoV-2 Wastewater Surveillance for Public Health Action. Emerg. Infect. Dis. J. 2021, 27, 1. [Google Scholar] [CrossRef]
- Lundy, L.; Fatta-Kassinos, D.; Slobodnik, J.; Karaolia, P.; Cirka, L.; Kreuzinger, N.; Castiglioni, S.; Bijlsma, L.; Dulio, V.; Deviller, G.; et al. Making Waves: Collaboration in the time of SARS-CoV-2—Rapid development of an international co-operation and wastewater surveillance database to support public health decision-making. Water Res. 2021, 199, 117167. [Google Scholar] [CrossRef]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098. [Google Scholar] [CrossRef]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E.; et al. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 3 August 2021).
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Gohl, D.M.; Garbe, J.; Grady, P.; Daniel, J.; Watson, R.H.B.; Auch, B.; Nelson, A.; Yohe, S.; Beckman, K.B. A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genomics 2020, 21, 863. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12, e02703–e02720. [Google Scholar] [CrossRef]
- Jahn, K.; Dreifuss, D.; Topolsky, I.; Kull, A.; Ganesanandamoorthy, P.; Fernandez-Cassi, X.; Bänziger, C.; Stachler, E.; Fuhrmann, L.; Jablonski, K.P.; et al. Detection of SARS-CoV-2 variants in Switzerland by genomic analysis of wastewater samples. MedRxiv 2021. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Munnink, B.B.O.; Schapendonk, C.M.E.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and diversity through community wastewater sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. J. 2021, 27, 1405. [Google Scholar] [CrossRef]
- Pérez-Cataluña, A.; Chiner-Oms, Á.; Cuevas-Ferrando, E.; Díaz-Reolid, A.; Falcó, I.; Randazzo, W.; Girón-Guzmán, I.; Allende, A.; Bracho, M.A.; Comas, I.; et al. Detection of genomic variants of SARS-CoV-2 circulating In wastewater by high-throughput sequencing. MedRxiv 2021. [Google Scholar] [CrossRef]
- Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eur. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.L.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.C.; Pinel, N.; May, P.; Wilmes, P. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016, 17, 260. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Popa, A.; Genger, J.-W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12, eabe2555. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, Snp.Eff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Ruden, D.; Cingolani, P.; Patel, V.; Coon, M.; Nguyen, T.; Land, S.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.M.; Aubrey, W.; De Grave, K.; Schietgat, L.; Creevey, C.J.; Clare, A. On the complexity of haplotyping a microbial community. Bioinformatics 2021, 37, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Kraberger, S.; Hadfield, J.; Driver, E.M.; Bowes, D.; Holland, L.A.; Faleye, T.O.C.; Adhikari, S.; Kumar, R.; Inchausti, R.; et al. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. Water Res. 2021, 205, 117710. [Google Scholar] [CrossRef]
- Sapoval, N.; Lou, E.; Hopkins, L.; Ensor, K.B.; Schneider, R.; Treangen, T.J.; Stadler, L.B. Enhanced Detection of Recently Emerged SARS-CoV-2 Variants of Concern in Wastewater. medRxiv 2021. [Google Scholar] [CrossRef]
- Rios, G.; Lacoux, C.; Leclercq, V.; Diamant, A.; Lebrigand, K.; Lazuka, A.; Soyeux, E.; Lacroix, S.; Fassy, J.; Couesnon, A.; et al. Monitoring SARS-CoV-2 variants alterations in Nice neighborhoods by wastewater nanopore sequencing. Lancet Reg. Health Eur. 2021. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Detected/Total Mutations | Found in Multiple Samples (>5) | Missense Variant | Synonymous Variant | Frameshift Variant | Inframe Deletion |
|---|---|---|---|---|---|---|
| ORF1ab | 660/2977 | 83/107 | 1281 (43%) | 659 (22%) | 896 (30%) | 48 (2%) |
| S | 141/578 | 21/55 | 264 (46%) | 109 (19%) | 182 (31%) | 7 (1%) |
| ORF3a | 77/165 | 11/11 | 103 (62%) | 24 (15%) | 31 (19%) | 5 (3%) |
| E | 11/49 | 0/0 | 20 (41%) | 8 (16%) | 13 (27%) | 1 (2%) |
| M | 36/126 | 10/11 | 38 (30%) | 37 (29%) | 38 (30%) | 1 (1%) |
| ORF6 | 11/49 | 1/1 | 14 (29%) | 13 (27%) | 18 (37%) | 2 (4%) |
| ORF7a | 31/68 | 3/3 | 34 (50%) | 12 (18%) | 18 (27%) | 1 (2%) |
| ORF7b | 12/34 | 2/2 | 15 (44%) | 8 (24%) | 9 (27%) | 1 (3%) |
| ORF8 | 41/97 | 10/13 | 49 (51%) | 18 (19%) | 21 (22%) | 1 (1%) |
| N | 100/287 | 23/28 | 145 (51%) | 64 (22%) | 66 (23%) | 5 (2%) |
| ORF10 | 0/2 | 0/0 | 0 (0%) | 0 (0%) | 1 (50%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herold, M.; d'Hérouël, A.F.; May, P.; Delogu, F.; Wienecke-Baldacchino, A.; Tapp, J.; Walczak, C.; Wilmes, P.; Cauchie, H.-M.; Fournier, G.; et al. Genome Sequencing of SARS-CoV-2 Allows Monitoring of Variants of Concern through Wastewater. Water 2021, 13, 3018. https://doi.org/10.3390/w13213018
Herold M, d'Hérouël AF, May P, Delogu F, Wienecke-Baldacchino A, Tapp J, Walczak C, Wilmes P, Cauchie H-M, Fournier G, et al. Genome Sequencing of SARS-CoV-2 Allows Monitoring of Variants of Concern through Wastewater. Water. 2021; 13(21):3018. https://doi.org/10.3390/w13213018
Chicago/Turabian StyleHerold, Malte, Aymeric Fouquier d'Hérouël, Patrick May, Francesco Delogu, Anke Wienecke-Baldacchino, Jessica Tapp, Cécile Walczak, Paul Wilmes, Henry-Michel Cauchie, Guillaume Fournier, and et al. 2021. "Genome Sequencing of SARS-CoV-2 Allows Monitoring of Variants of Concern through Wastewater" Water 13, no. 21: 3018. https://doi.org/10.3390/w13213018