
Ascomycetes versus Spent Mushroom Substrate in Mycoremediation of Dredged Sediments Contaminated by Total Petroleum Hydrocarbons: The Involvement of the Bacterial Metabolism

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. TPH Contaminated Dredged Sediment: Sediment, Fungal Strain, Diesel Oil and Chemicals
2.2. Mesocosms-Scale Experimentation
2.3. Metabarcoding Analysis
2.4. Data Analysis
2.5. Quantification of Total Petroleum Hydrocarbons, Humic Acid, Fulvic Acid, and Ergosterol in Soil Samples
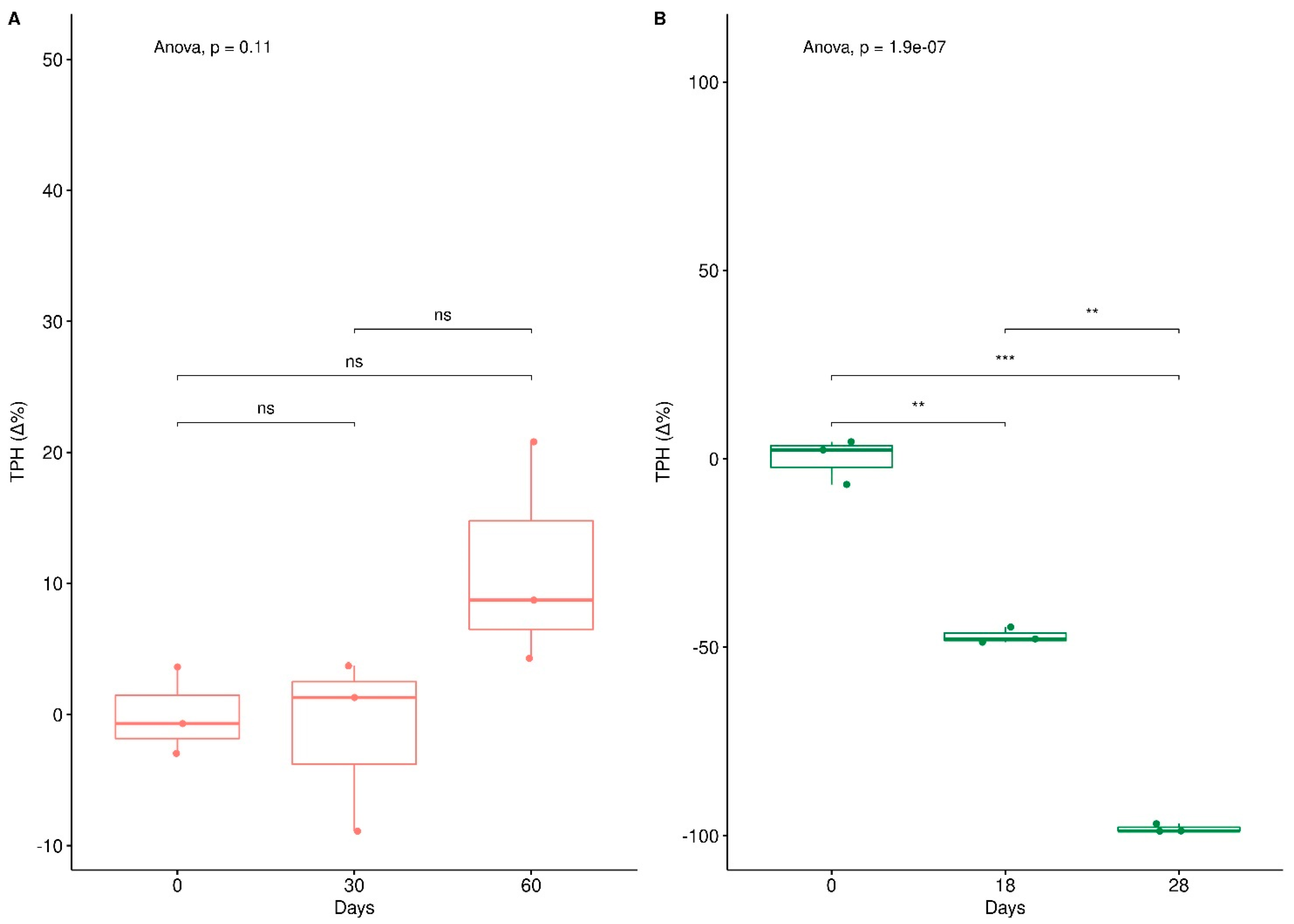
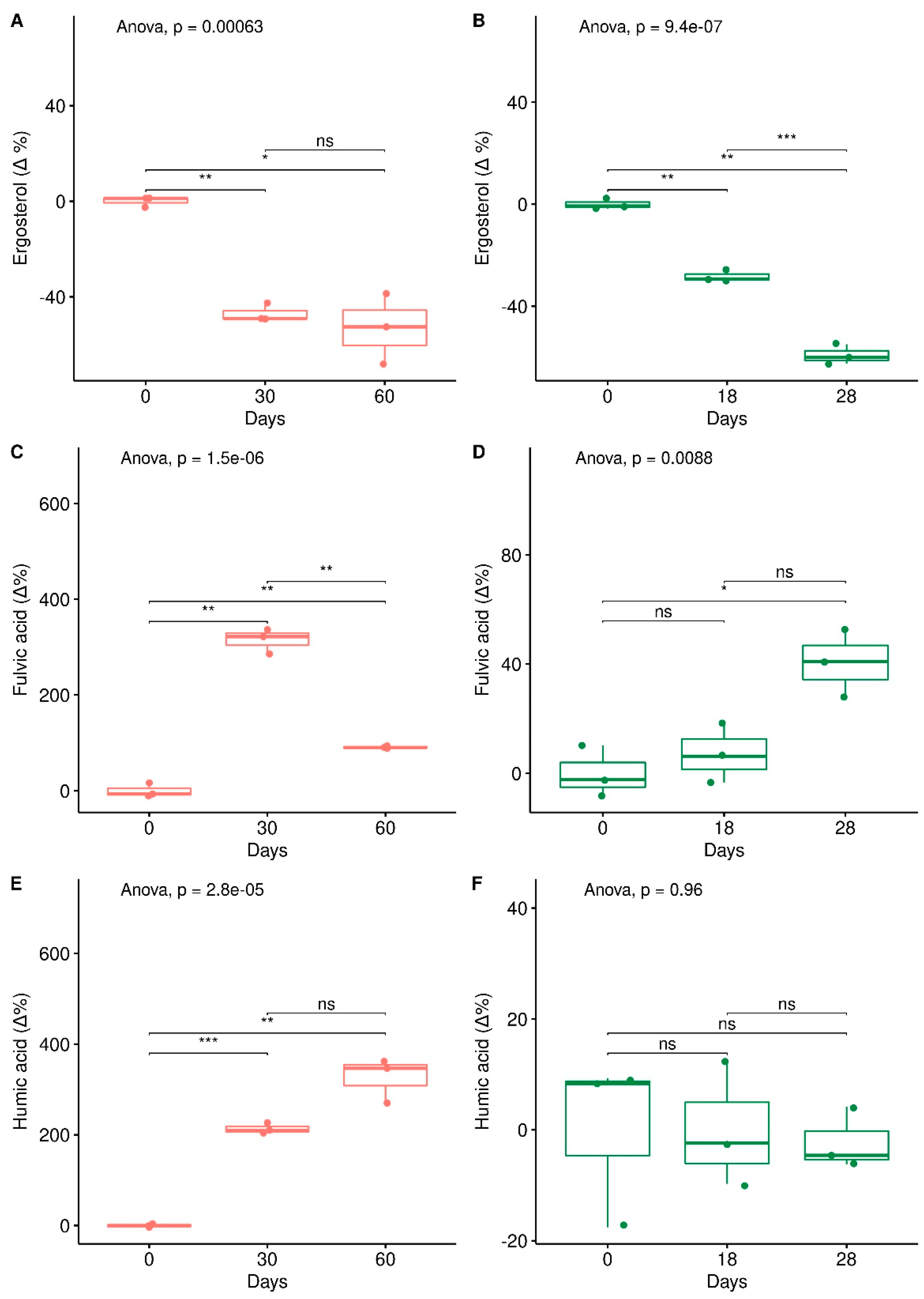
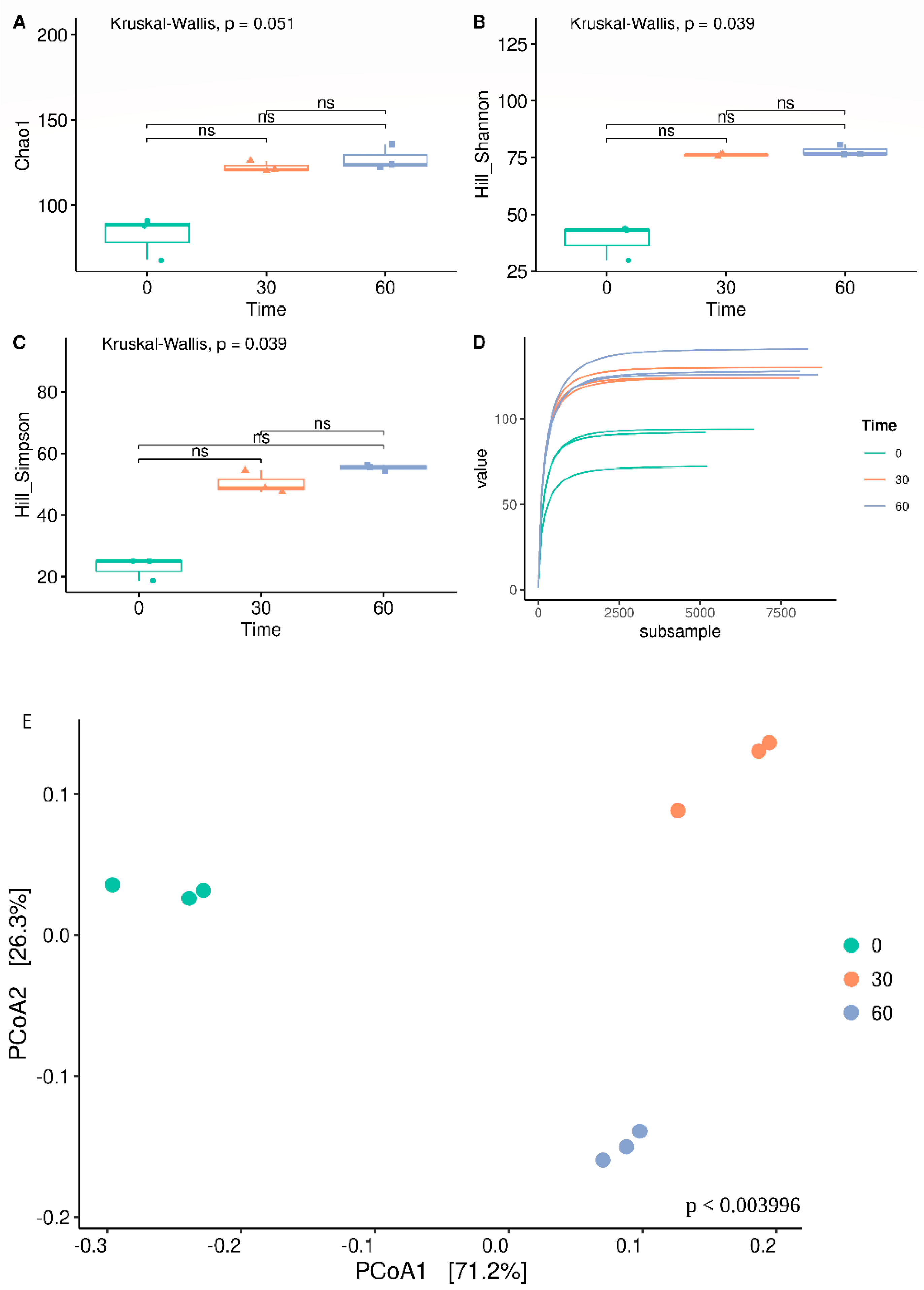
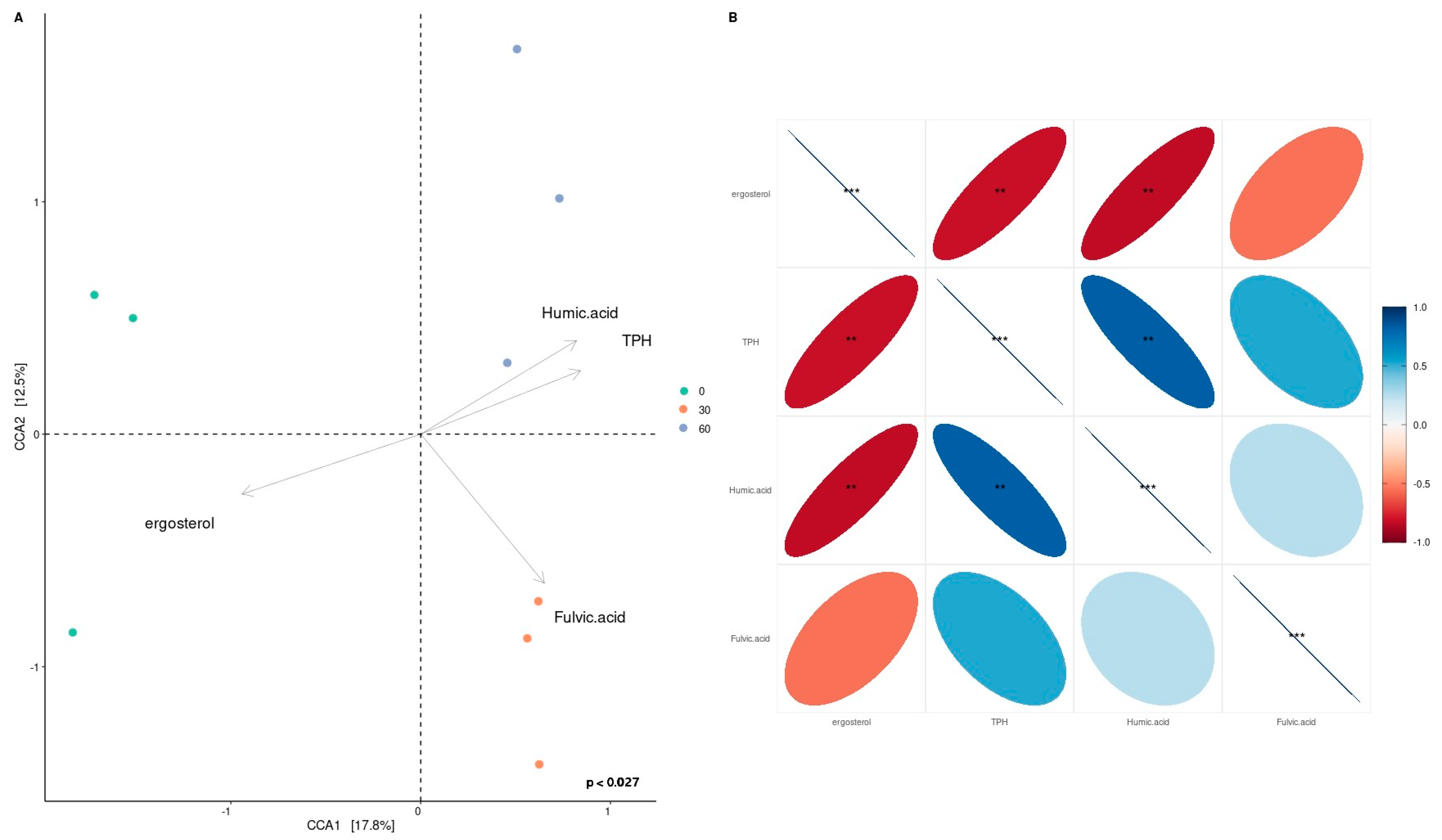
3. Results
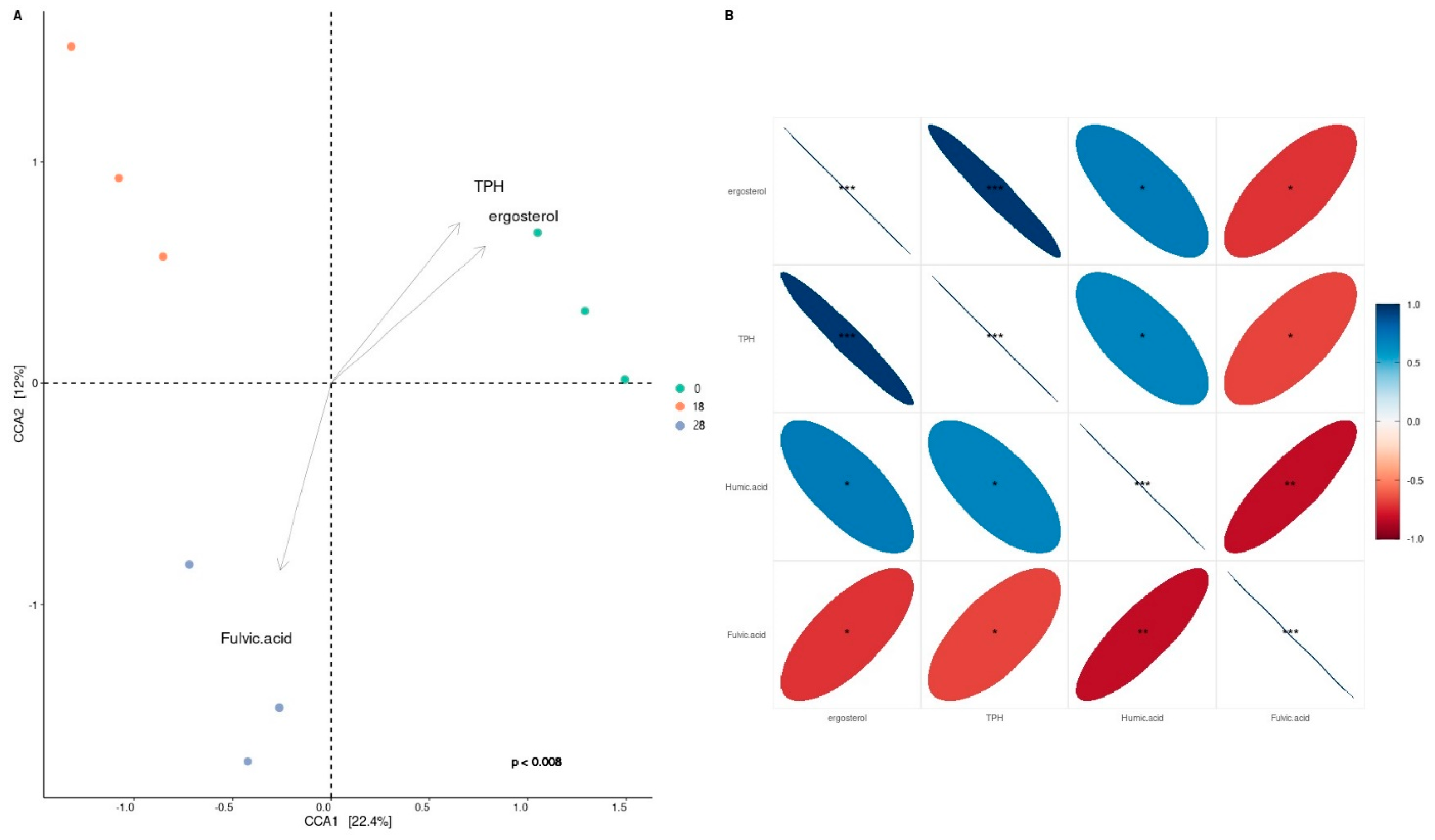
3.1. Depletion of Total Petroleum Hydrocarbons and Humic and Fulvic Acid and Ergosterol Content in Mesocosms
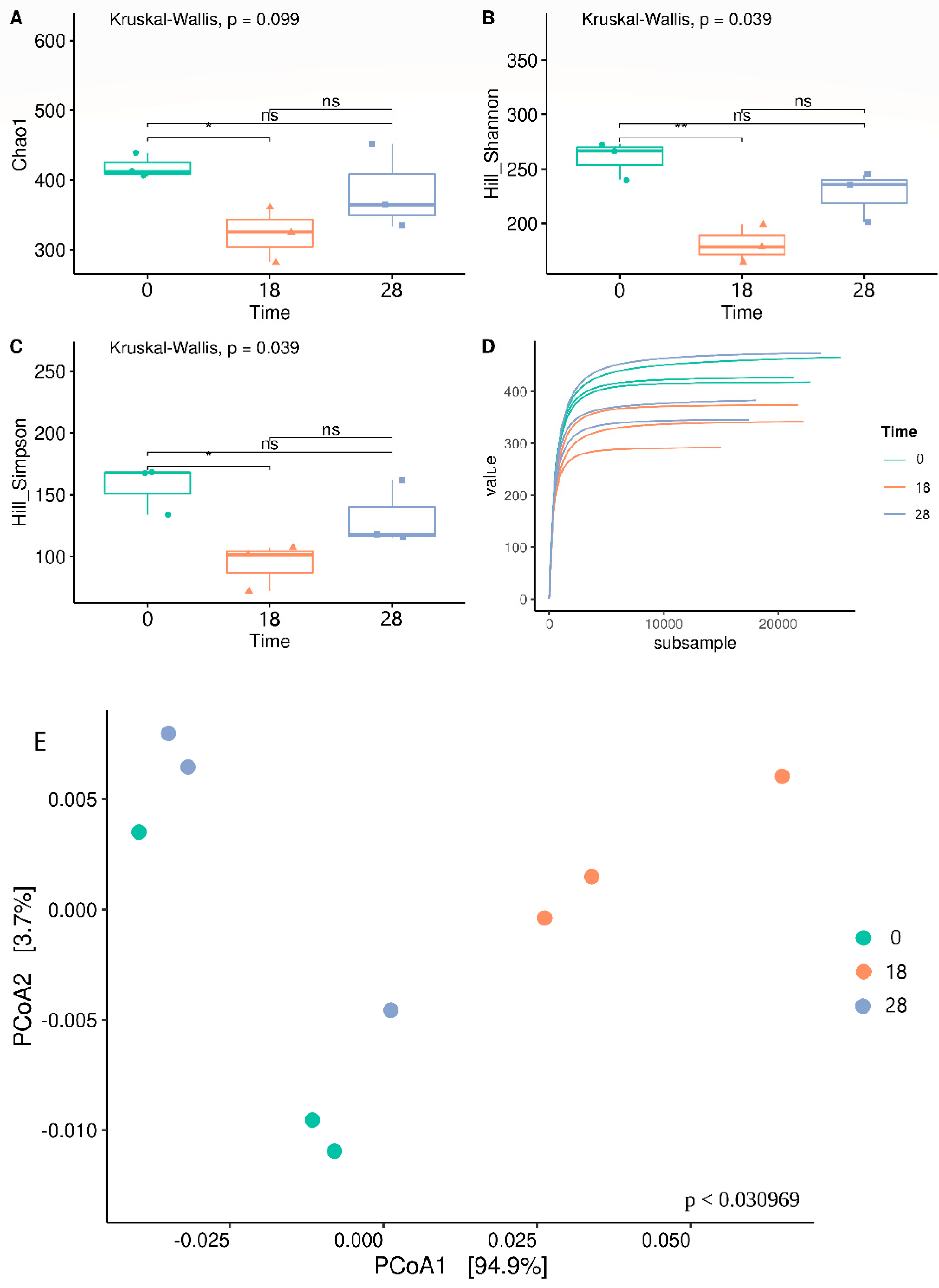
3.2. Bacterial Biodiversity
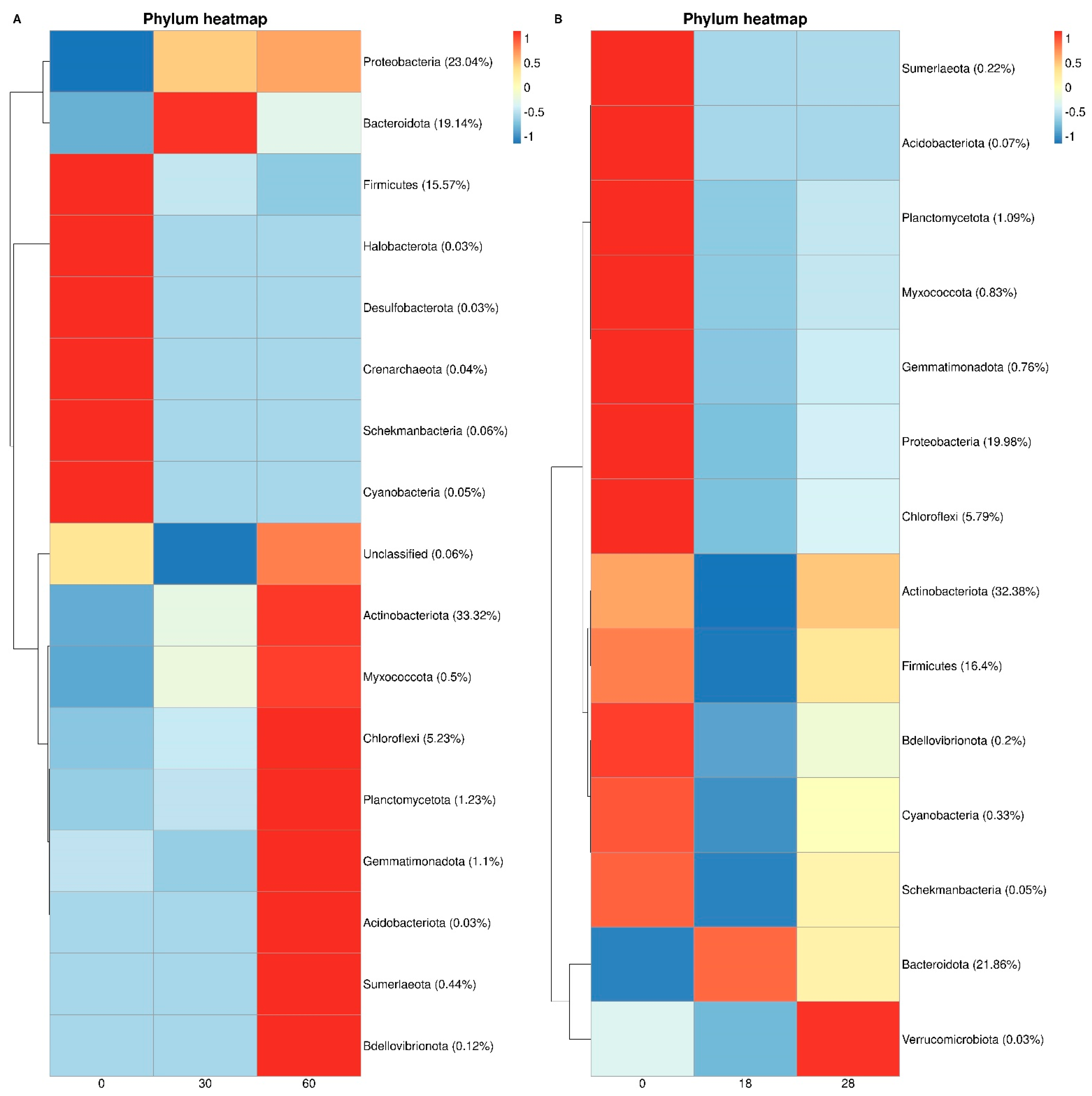
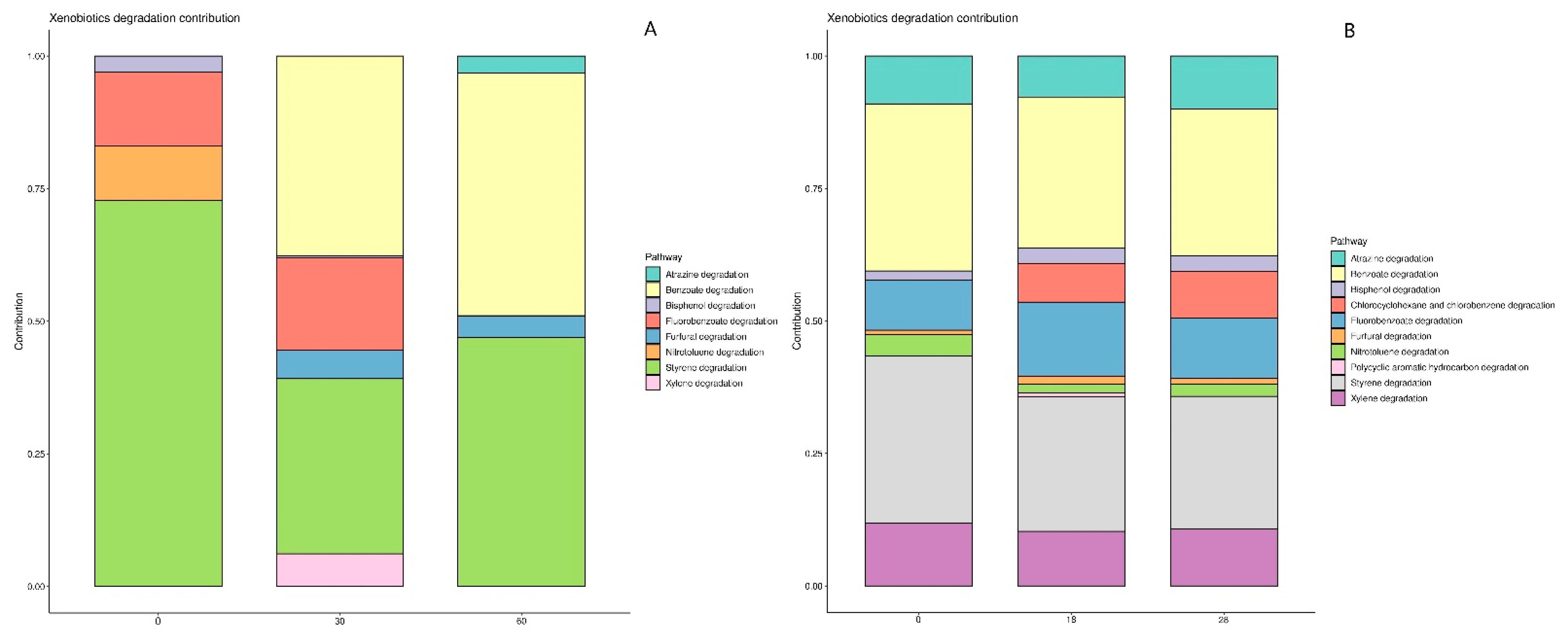
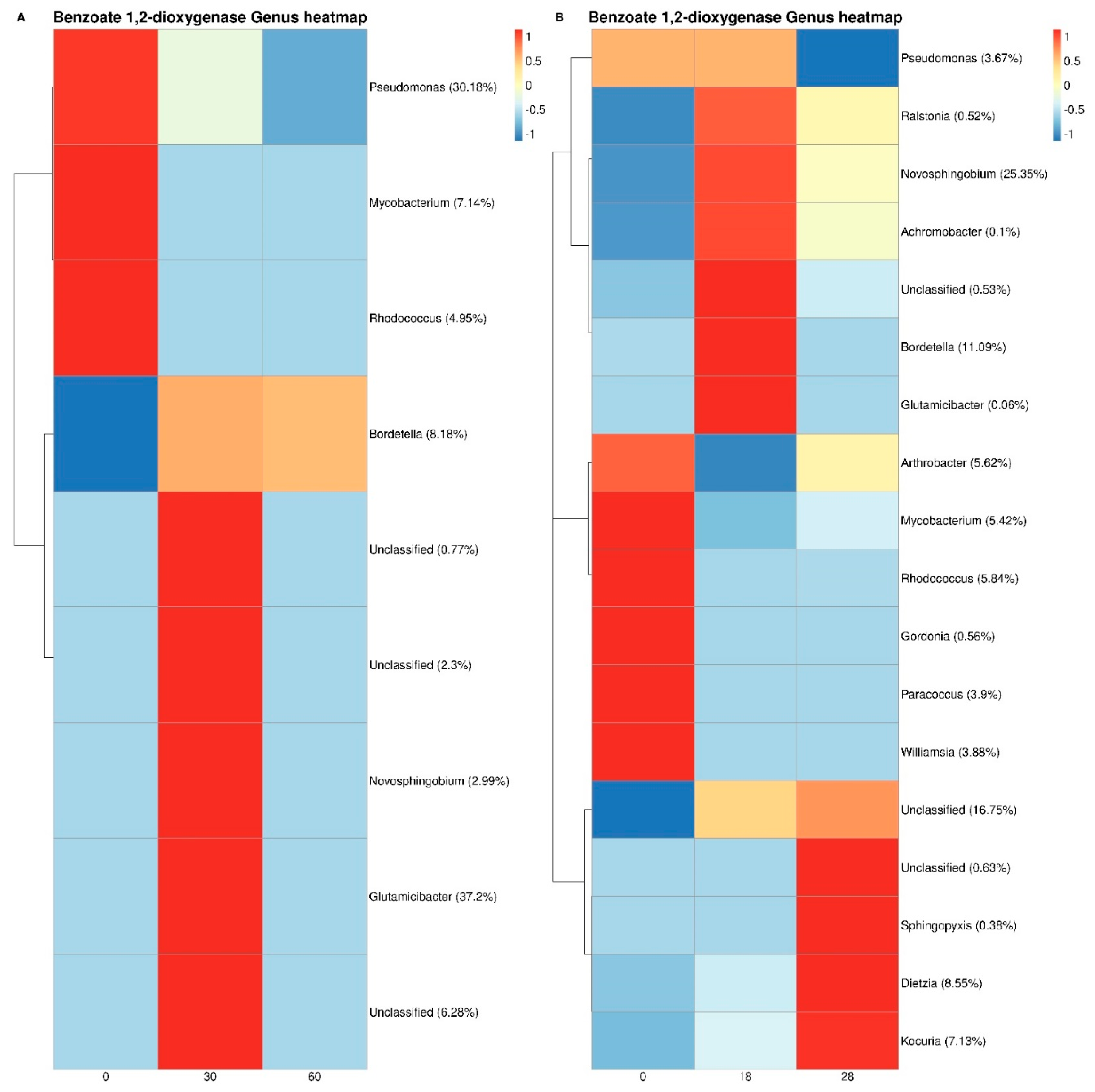
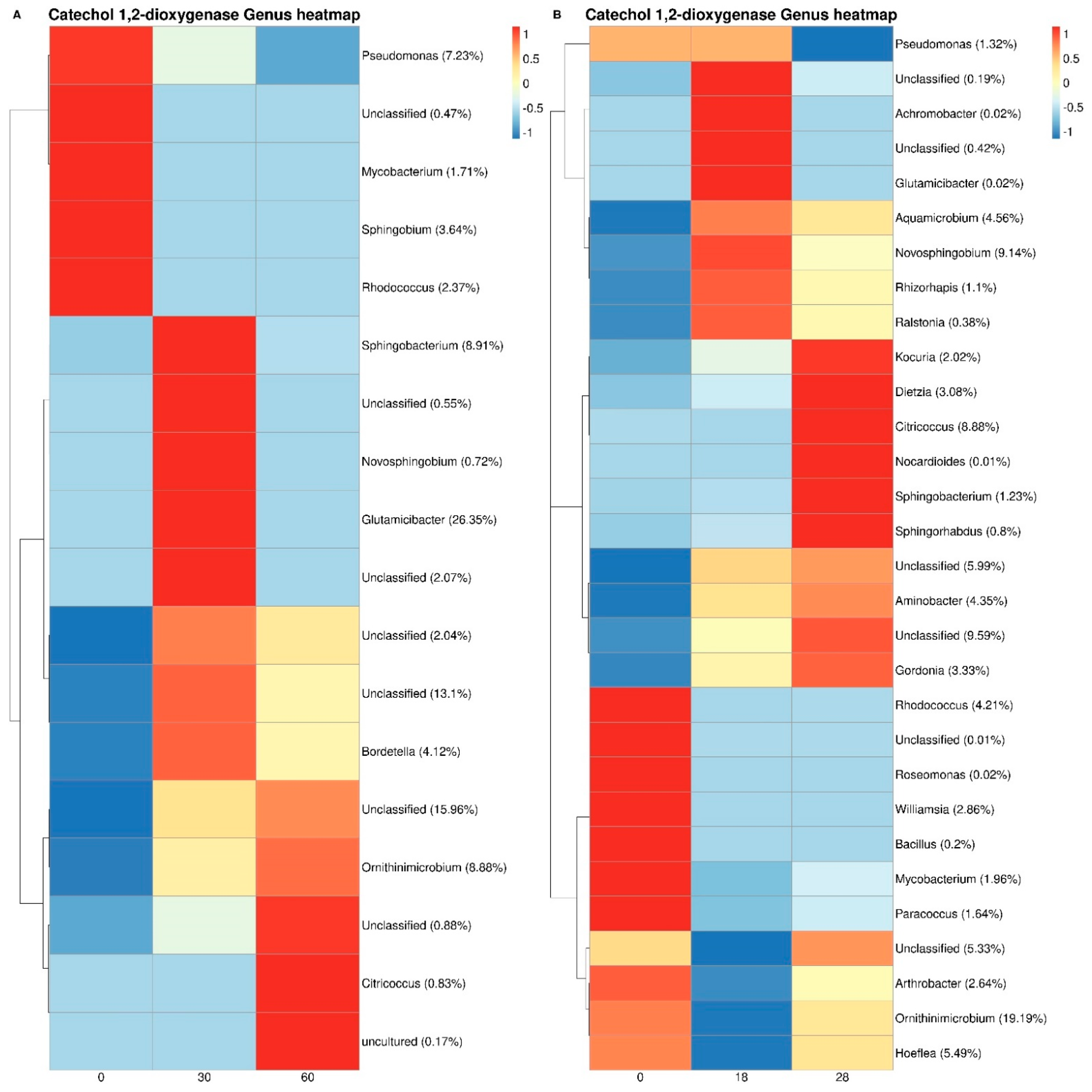
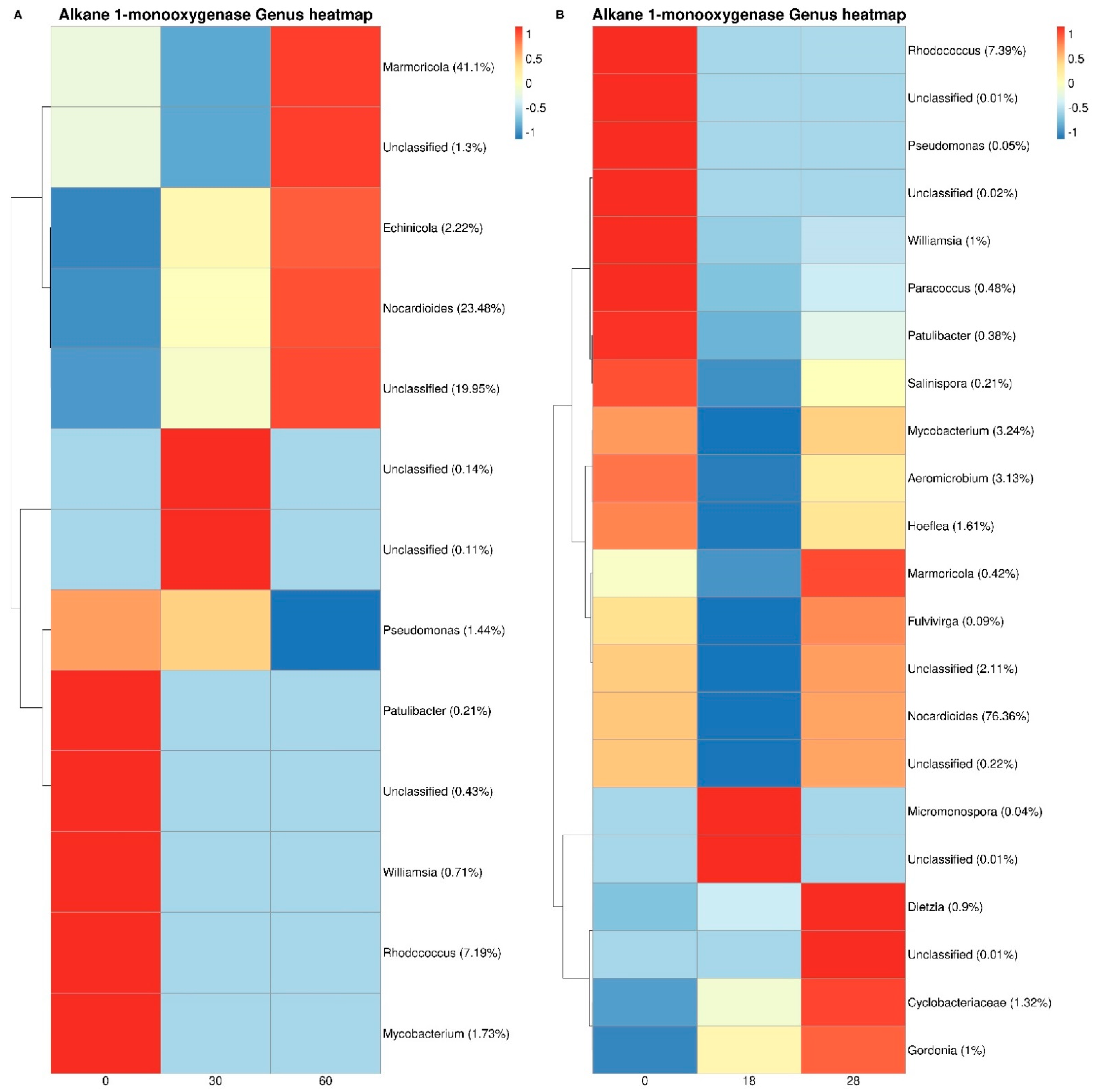
3.3. Bacterial Taxonomy and Predictive Functional Metagenomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- European Sediment Network. In Proceedings of the 9th International SedNet Conference, Kraków, Poland, 23–26 September 2015.
- Wang, M.; Wang, C.; Hu, X.; Zhang, H.; He, S.; Lv, S. Distributions and sources of petroleum, aliphatic hydrocarbons and polycyclic aromatic hydrocarbons (PAHs) in surface sediments from Bohai Bay and its adjacent river, China. Mar. Pollut. Bull. 2015, 90, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.M.; Scarlett, A.G.; Lewis, C.A.; Belt, S.T.; Rowland, S.J. Unresolved complex mixtures (UCMs) of aromatic hydrocarbons: Branched alkyl indanes and branched alkyl tetralins are present in UCMs and accumulated by and toxic to the mussel Mytilus edulis. Environ. Sci. Technol. 2008, 42, 8122–8126. [Google Scholar] [CrossRef] [PubMed]
- Federici, E.; Giubilei, M.A.; Covino, S.; Zanaroli, G.; Fava, F.; D’Annibale, A.; Petruccioli, M. Addition of maize stalks and soybean oil to a historically PCB-contaminated soil: Effect on degradation performance and indigenous microbiota. New Biotechnol. 2012, 30, 69–79. [Google Scholar] [CrossRef]
- Maila, M.P.; Cloete, T.E. Bioremediation of petroleum hydrocarbons through landfarming: Are simplicity and cost-effectiveness the only advantages? Rev. Environ. Sci. Biotechnol. 2004, 3, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Becarelli, S.; Chicca, I.; Siracusa, G.; La China, S.; Gentini, A.; Lorenzi, R.; Munz, G.; Petroni, G.; Levin, D.B.; Di Gregorio, S. Hydrocarbonoclastic Ascomycetes to enhance co-composting of total petroleum hydrocarbon (TPH) contaminated dredged sediments and lignocellulosic matrices. New Biotechnol. 2019, 50, 27–36. [Google Scholar] [CrossRef]
- Grard, B.J.; Manouchehri, N.; Aubry, C.; Frascaria-Lacoste, N.; Chenu, C. Potential of Technosols Created with Urban By-Products for Rooftop Edible Production. Int. J. Environ. Res. Public Health. 2020, 17, 3210. [Google Scholar] [CrossRef] [PubMed]
- Becher, M.; Banach-Szott, M.; Godlewska, A. Organic Matter Properties of Spent Button Mushroom Substrate in the Context of Soil Organic Matter Reproduction. Agronomy 2021, 11, 204. [Google Scholar] [CrossRef]
- Chiu, S.W.; Law, S.C.; Ching, M.L.; Cheung, K.W.; Chen, M.J. Themes for mushroom exploitation in the 21st century: Sustainability, waste management and conservation. J. Gen. Appl. Microbiol. 2000, 46, 269–282. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, S.; Becarelli, S.; Siracusa, G.; Riffini Castiglione, M.; Petroni, G.; Masini, G.; Gentini, A.; Rubia de Lima e Silva, M.; Lorenzi, R. Pleurotus ostreatus spent mushroom substrate for the degradation of polycyclic aromatic hydrocarbons: The case study of a pilot dynamic biopile for the decontamination of a historically contaminated soil. J. Chem. Technol. Biotechnol. 2016, 91, 1654–1664. [Google Scholar] [CrossRef]
- Garcia-Delgado, C.; D’Annibale, A.; Pesciaroli, L.; Yunta, F.; Crognale, S.; Petruccioli, M.; Eymar, E. Implications of polluted soil biostimulation and bioaugmentation with spent mushroom substrate (Agaricus bisporus) on the microbial community and polycyclic aromatic hydrocarbons biodegradation. Sci. Total Environ. 2015, 508, 20–28. [Google Scholar] [CrossRef]
- Siracusa, G.; Becarelli, S.; Lorenzi, R.; Gentini, A.; Di Gregorio, S. PCB in the environment: Bio-based processes for soil decontamination and management of waste from the industrial production of Pleurotus ostreatus. New Biotechnol. 2017, 25 Pt B, 232–239. [Google Scholar] [CrossRef]
- Marín-Benito, J.M.; Sánchez-Martín, M.J.; Rodríguez-Cruz, M.S. Impact of Spent Mushroom Substrates on the Fate of Pesticides in Soil, and Their Use for Preventing and/or Controlling Soil and Water Contamination: A Review. Toxics 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buiarelli, F.; Canepari, S.; Di Filippo, P.; Perrino, C.; Pomata, D.; Riccardi, C.; Speziale, R. Extraction and analysis of fungal spore biomarkers in atmospheric bioaerosol by HPLC-MS-MS and GC-MS. Talanta 2013, 105, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Olsson, P.A.; Larsson, L.; Bago, B.; Wallander, H.; Van Aarle, I.M. Ergosterol and fatty acids for biomass estimation of mycorrhizal fungi. New Phytol. 2003, 159, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, D.J.; Gakhar, L.; Ramaswamy, S. Rieske business: Structure-function of Rieske non-heme oxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 175–190. [Google Scholar] [CrossRef]
- Shen, F.T.; Young, L.S.; Hsieh, M.F.; Lin, S.Y.; Young, C.C. Molecular detection and phylogenetic analysis of the alkane 1-monooxygenase gene from Gordonia spp. Syst. Appl. Microbiol. 2010, 33, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Doni, S.; Macci, C.; Peruzzi, E.; Iannelli, R.; Ceccanti, B.; Masciandaro, G. Decontamination and functional reclamation of dredged brackish sediments. Biodegradation 2013, 24, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Ugolini, F.; Mariotti, B.; Maltoni, A.; Tani, A.; Salbitano, F.; Izquierdo, C.G.; Macci, C.; Masciandaro, G.; Tognetti, R. A tree from waste: Decontaminated dredged sediments for growing forest tree seedlings. J. Environ. Manag. 2018, 211, 269–277. [Google Scholar] [CrossRef]
- Becher, M. Properties of organic matter of soil fertilised with spent mushroom (Agaricus, L.) Substrate. Acta Agroph. 2013, 20, 241–252. [Google Scholar]
- Zied, D.C.; Sánchez, J.E.; Noble, R.; Pardo-Giménez, A. Use of Spent Mushroom Substrate in New Mushroom Crops to Promote the Transition towards A Circular Economy. Agronomy 2020, 10, 1239. [Google Scholar] [CrossRef]
- Paredes, C.; Medina, E.; Bustamante, M.A.; Moral, R. Effects of spent mushroom substrates and inorganic fertilizer on the characteristics of a calcareous clayey-loam soil and lettuce production. Soil Use Manag. 2016, 32, 487–494. [Google Scholar] [CrossRef]
- Medina, E.; Paredes, C.; Bustamante, M.A.; Moral, R.; Moreno-Caselles, J. Relationships between soil physico-chemical, chemical and biological properties in a soil amended with spent mushroom substrate. Geoderma 2012, 173, 152–161. [Google Scholar] [CrossRef]
- Hanafi, F.H.; Rezania, S.; Taib, S.M.; Din, M.F.; Yamauchi, M.; Sakamoto, M.; Hara, H.; Park, J.; Ebrahimi, S.S. Environmentally sustainable applications of agro-based spent mushroom substrate (SMS): An overview. J. Mater. Cycles Waste Manag. 2018, 20, 1383–1396. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Wang, Y.; Zhao, Y.; Wei, Z.; Zhao, M.; Song, C.; Liu, Y. Nitrate shifted microenvironment: Driven aromatic-ring cleavage microbes and aromatic compounds precursor biodegradation during sludge composting. Bioresour. Technol. 2021, 342, 125907. [Google Scholar] [CrossRef] [PubMed]
- Becarelli, S.; Chicca, I.; La China, S.; Siracusa, G.; Bardi, A.; Gullo, M.; Petroni, G.; Levin, D.B.; Di Gregorio, S. A New Ciboria sp. for Soil Mycoremediation and the Bacterial Contribution to the Depletion of Total Petroleum Hydrocarbons. Front. Microbiol. 2021, 12, 647373. [Google Scholar] [CrossRef]
- Hammershøj, R.; Birch, H.; Redman, A.D.; Mayer, P. Mixture Effects on Biodegradation Kinetics of Hydrocarbons in Surface Water: Increasing Concentrations Inhibited Degradation whereas Multiple Substrates Did Not. Environ. Sci. Technol. 2019, 53, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Knightes, C.D.; Peters, C.A. Multisubstrate biodegradation kinetics for binary and complex mixtures of polycyclic aromatic hydrocarbons. Environ. Toxicol. Chem. 2006, 25, 1746–1756. [Google Scholar] [CrossRef]
- Wang, M.; Garrido-Sanz, D.; Sansegundo-Lobato, P.; Redondo-Nieto, M.; Conlon, R.; Martin, M.; Mali, R.; Liu, X.; Dowling, D.N.; Rivilla, R.; et al. Soil Microbiome Structure and Function in Ecopiles Used to Remediate Petroleum-Contaminated Soil. Front. Environ. Sci. 2021, 9, 624070. [Google Scholar] [CrossRef]
- Lu, Y.; Zheng, G.; Zhou, W.; Wang, J.; Zhou, L. Bioleaching conditioning increased the bioavailability of polycyclic aromatic hydrocarbons to promote their removal during co-composting of industrial and municipal sewage sludges. Sci. Total Environ. 2019, 665, 1073–1082. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wang, B.J.; Jiang, C.Y.; Liu, S.J. Ornithinimicrobium pekingense sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 2008, 58 Pt 1, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Guo, S.; Hartog, N.; Yuan, Y.; Yang, X. Isolation and characterization of heavy polycyclic aromatic hydrocarbon-degrading bacteria adapted to electrokinetic conditions. Biodegradation 2016, 27, 1–13. [Google Scholar] [CrossRef]
- Chettri, B.; Singh, A.K. Kinetics of hydrocarbon degradation by a newly isolated heavy metal tolerant bacterium Novosphingobium panipatense P5:ABC. Bioresour. Technol. 2019, 294, 122190. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, C.W.; Kitts, C.L. Bacterial succession in a petroleum land treatment unit. Appl. Environ. Microbiol. 2004, 70, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzeszcz, J.; Kapusta, P.; Steliga, T.; Turkiewicz, A. Hydrocarbon Removal by Two Differently Developed Microbial Inoculants and Comparing Their Actions with Biostimulation Treatment. Molecules 2020, 25, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.B.; Singh, S.; Patel, A.; Jain, K.; Amin, S.; Madamwar, D. Synergistic biodegradation of phenanthrene and fluoranthene by mixed bacterial cultures. Bioresour. Technol. 2019, 284, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Gran-Scheuch, A.; Ramos-Zuñiga, J.; Fuentes, E.; Bravo, D.; Pérez-Donoso, J.M. Effect of Co-contamination by PAHs and Heavy Metals on Bacterial Communities of Diesel Contaminated Soils of South Shetland Islands, Antarctica. Microorganisms 2020, 8, 1749. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Dredged Sediment |
|---|---|
| Total Phosphate | 12 ± 0.1 mg/Kg |
| Total Nitrogen | 0.29 ± 0.03 mg/Kg |
| Chloride | 39 ± 0.3 g/L |
| TPH C > 12 | 2864 ± 99 mg/Kg |
| pH | 8.2 |
| Granulometric fraction | <2 mm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becarelli, S.; Siracusa, G.; Chicca, I.; Bernabei, G.; Di Gregorio, S. Ascomycetes versus Spent Mushroom Substrate in Mycoremediation of Dredged Sediments Contaminated by Total Petroleum Hydrocarbons: The Involvement of the Bacterial Metabolism. Water 2021, 13, 3040. https://doi.org/10.3390/w13213040
Becarelli S, Siracusa G, Chicca I, Bernabei G, Di Gregorio S. Ascomycetes versus Spent Mushroom Substrate in Mycoremediation of Dredged Sediments Contaminated by Total Petroleum Hydrocarbons: The Involvement of the Bacterial Metabolism. Water. 2021; 13(21):3040. https://doi.org/10.3390/w13213040
Chicago/Turabian StyleBecarelli, Simone, Giovanna Siracusa, Ilaria Chicca, Giacomo Bernabei, and Simona Di Gregorio. 2021. "Ascomycetes versus Spent Mushroom Substrate in Mycoremediation of Dredged Sediments Contaminated by Total Petroleum Hydrocarbons: The Involvement of the Bacterial Metabolism" Water 13, no. 21: 3040. https://doi.org/10.3390/w13213040