
Community Composition and Function of Bacteria in Activated Sludge of Municipal Wastewater Treatment Plants

 and
and
Abstract
:1. Introduction
2. Materials and Methods
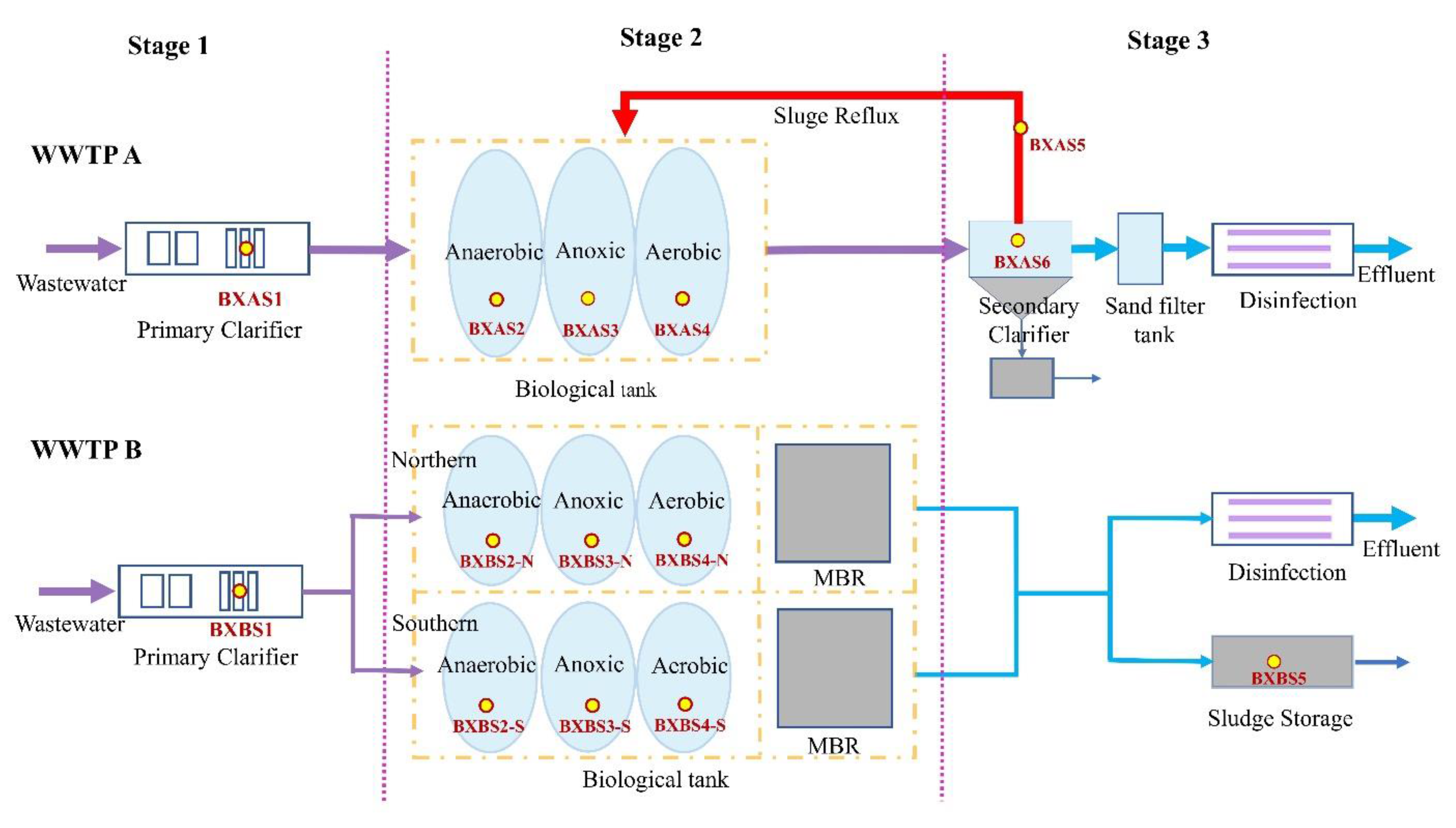
2.1. Sample Collection
2.2. Physical and Chemical Analysis
2.3. DNA Extraction and Illumina NovaSeq Sequencing
2.4. Data Processing and Analysis
2.5. Community Structure Analysis
3. Results
3.1. Sample Characteristics
3.2. Richness and Diversity of Bacterial Communities
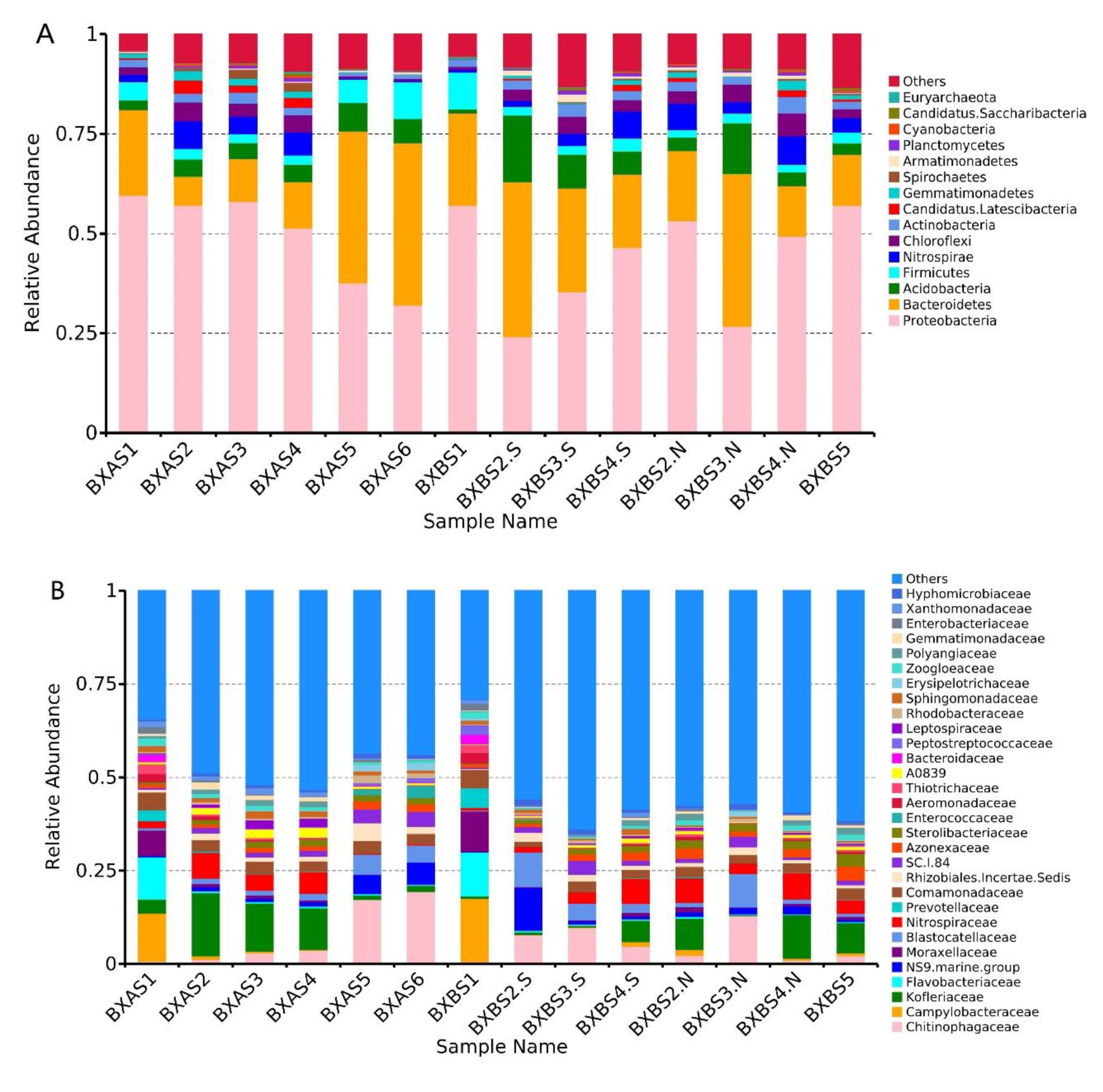
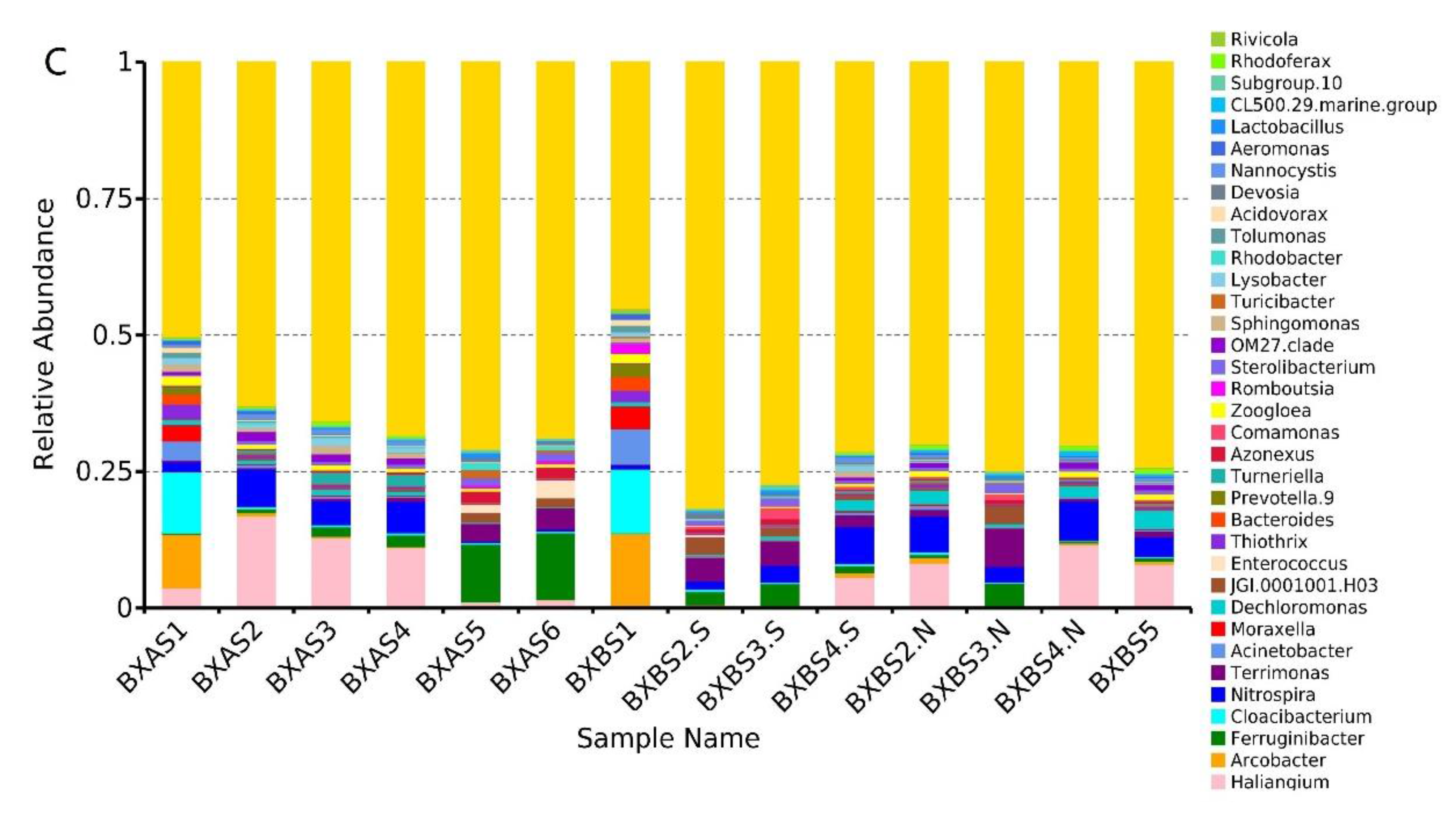
3.3. Microbial Taxonomy and Community Composition Analysis
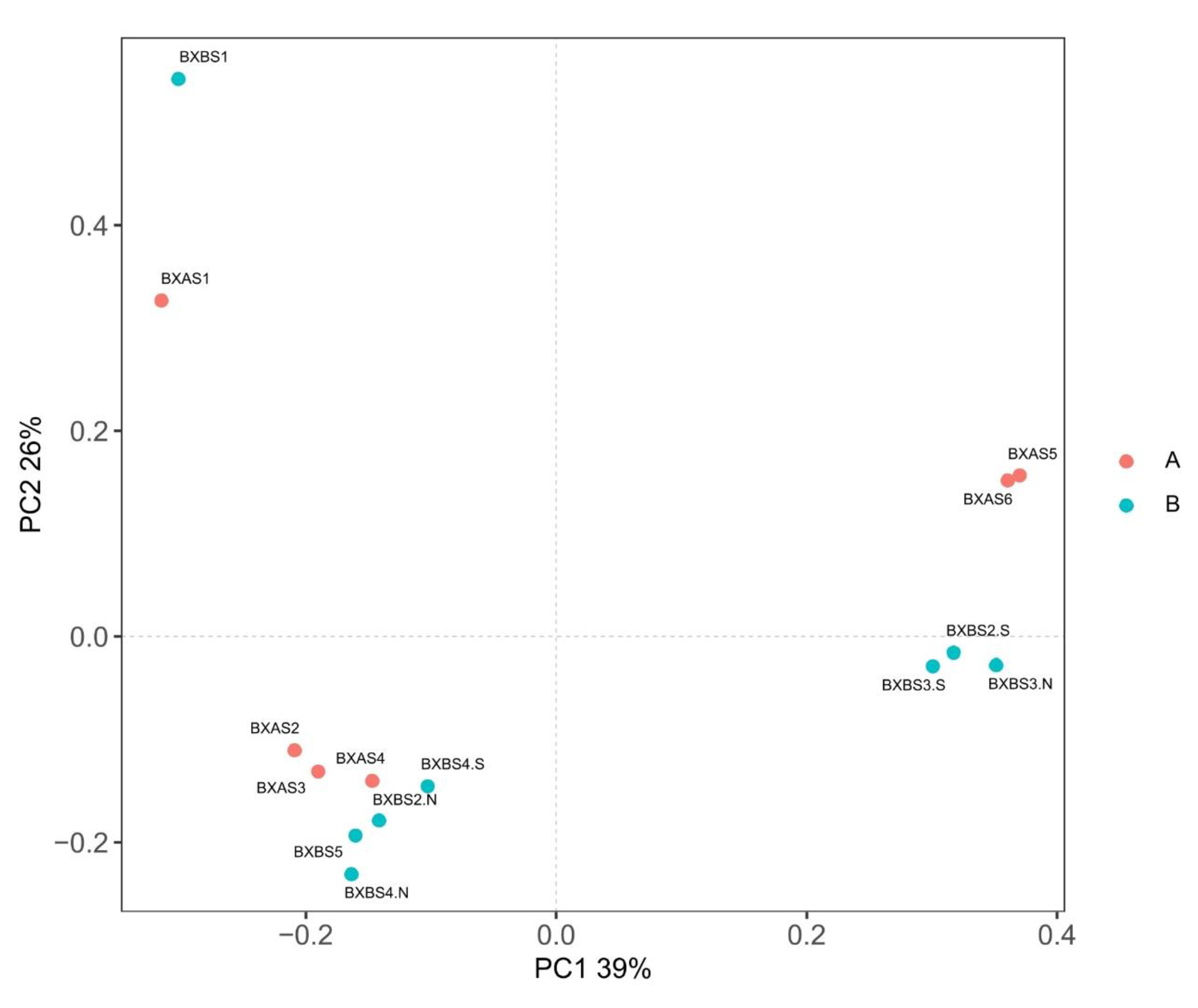
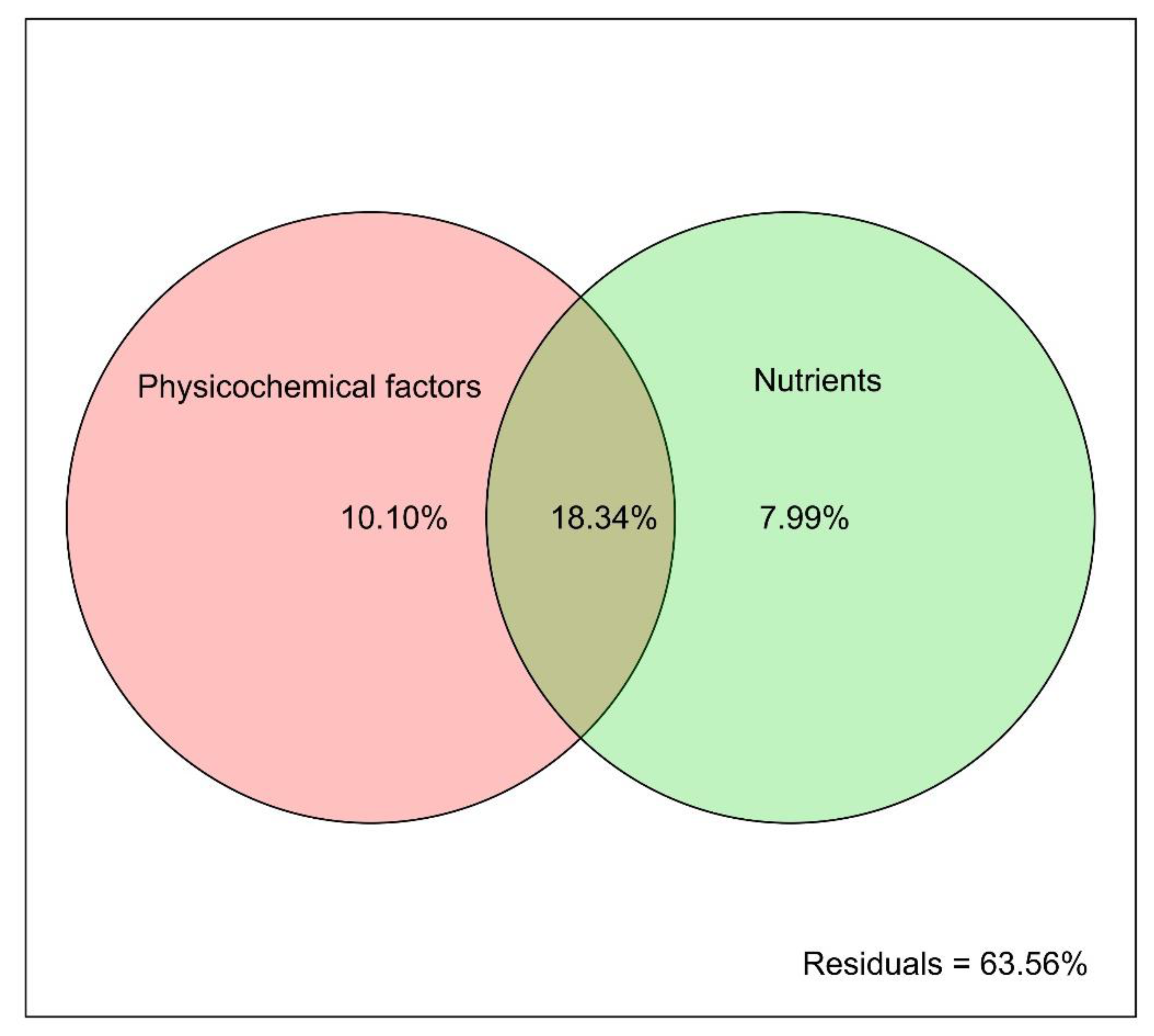
3.4. Microbial Community Variation Analysis
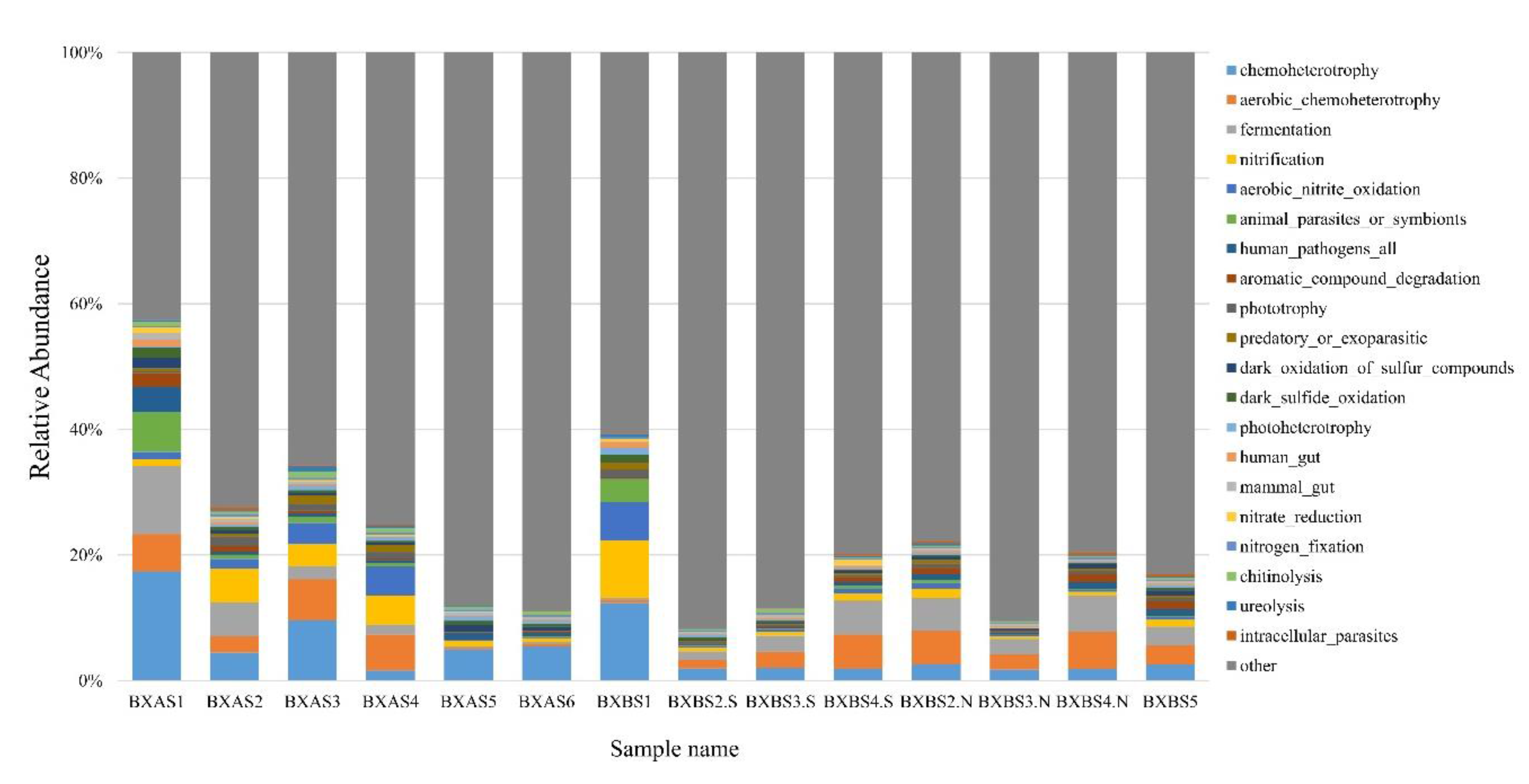
3.5. Prediction of Microbial Community Function
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolff, S.; Weber, F.; Kerpen, J.; Winklhofer, M.; Engelhart, M.; Barkmann, L. Elimination of microplastics by downstream sand filters in wastewater treatment. Water 2021, 13, 33. [Google Scholar] [CrossRef]
- Paliaga, P.; Felja, I.; Budisa, A.; Ivancic, I. The impact of a fish cannery wastewater discharge on the bacterial community structure and sanitary conditions of marine coastal sediments. Water 2019, 11, 2566. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Karim, M.R.; Zheng, X.; Li, X. Heavy metal and metalloid pollution of soil, water and foods in bangladesh: A critical review. Environ. Res. Public Health 2018, 15, 2825. [Google Scholar] [CrossRef] [Green Version]
- Agoro, M.A.; Adeniji, A.O.; Adefisoye, M.A.; Okoh, O.O. Heavy metals in wastewater and sewage sludge from selected municipal treatment plants in eastern cape province, south Africa. Water 2020, 12, 2746. [Google Scholar] [CrossRef]
- Wang, M.; Wu, Y.; Yang, B.; Deng, P.; Zhong, Y.; Fu, C.; Lu, Z.; Zhang, P.; Wang, J.; Qu, Y. Comparative study of the effect of rice husk-based powders used as physical conditioners on sludge dewatering. Sci. Rep. 2020, 10, 17230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shao, M.-F.; Ye, L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012, 6, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Ju, F.; Zhang, T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 2015, 9, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, Z.; Fang, W.; Gao, G. Composition of bacterial communities in municipal wastewater treatment plant. Sci. Total Environ. 2019, 689, 1181–1191. [Google Scholar] [CrossRef]
- Nielsen, P.H.; Saunders, A.M.; Hansen, A.A.; Larsen, P.; Nielsen, J.L. Microbial communities involved in enhanced biological phosphorus removal from wastewater—a model system in environmental biotechnology. Curr. Opin. Biotechnol. 2012, 23, 452–459. [Google Scholar] [CrossRef]
- Ju, F.; Guo, F.; Ye, L.; Xia, Y.; Zhang, T. Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ. Microbiol. Rep. 2014, 6, 80–89. [Google Scholar] [CrossRef]
- Daims, H.; Taylor, M.W.; Wagner, M. Wastewater treatment: A model system for microbial ecology. Trends Biotechnol. 2006, 24, 483–489. [Google Scholar] [CrossRef]
- Wan, W.; He, D.; Xue, Z. Removal of nitrogen and phosphorus by heterotrophic nitrification-aerobic denitrification of a denitrifying phosphorus-accumulating bacterium Enterobacter cloacae hw-15. Ecol. Eng. 2017, 99, 199–208. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, L.; Liu, L.; Su, C.; Dou, L.; Su, Z.; He, Z. Phosphorus and nitrogen removal by a novel phosphate-accumulating organism, Arthrobacter sp. Hhep5 capable of heterotrophic nitrification-aerobic denitrification: Safety assessment, removal characterization, mechanism exploration and wastewater treatment. Bioresour. Technol. 2020, 312, 123633. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Thill, A.; Purkhold, U.; Walcher, M.; Bottero, J.Y.; Ginestet, P.; Nielsen, P.H.; Wuertz, S.; Wagner, M. Characterization of activated sludge flocs by confocal laser scanning microscopy and image analysis. Water Res. 2003, 37, 2043–2052. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, T. Profiling bulking and foaming bacteria in activated sludge by high throughput sequencing. Water Res. 2012, 46, 2772–2782. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Zhang, T.; Wang, T.; Fang, Z. Microbial structures, functions, and metabolic pathways in wastewater treatment bioreactors revealed using high-throughput sequencing. Environ. Sci. Technol. 2012, 46, 13244–13252. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, X.; Zhu, L. Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 2017, 7, 17930. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Mao, Y.; Xu, J.; Tian, L.; Wang, Y.; Iqbal, W.; Yang, B.; Liu, C.; Zhao, X.; Wang, Y. Characterizing community dynamics and exploring bacterial assemblages in two activated sludge systems. Appl. Microbiol. Biotechnol. 2020, 104, 1795–1808. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Lu, Y.; Zhu, G.; Cheng, H. Simultaneous nitrification, denitrification and phosphorus removal (SNDPR) at low atmosphere pressure. Biochem. Eng. J. 2020, 160, 107629. [Google Scholar] [CrossRef]
- American Public Health Association. Standard Methods for the Examination of Water and Wastewater; APHA: Washington, DC, USA, 2007. [Google Scholar]
- Liu, M.; Gill, J.J.; Young, R.; Summer, E.J. Bacteriophages of wastewater foaming-associated filamentous Gordonia reduce host levels in raw activated sludge. Sci. Rep. 2015, 5, 13754. [Google Scholar] [CrossRef]
- Justin, K.; Jesse, S.; Anton, W.W.; Antonio, G.; Gregory, C.J.; Rob, K. Using QIIME to analyze 16s rRNA gene sequences from microbial communities. Curr. Protoc. 2012, 27, 1E.5.1–1E.5.20. [Google Scholar]
- Magoc, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.-A.; Balint, M.; Greshake, B.; Bandow, C.; Römbke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 2013, 65, 128–132. [Google Scholar] [CrossRef]
- Somerfield, P.J. Identification of the Bray-Curtis similarity index: Comment on Yoshioka (2008). Mar. Ecol. Prog. Ser. 2008, 372, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, P.M. Misidentification of the Bray-Curtis similarity index. Mar. Ecol. Prog. Ser. 2008, 368, 309–310. [Google Scholar] [CrossRef] [Green Version]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Pote, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef]
- Gao, P.; Xu, W.; Sontag, P.; Li, X.; Xue, G.; Liu, T.; Sun, W. Correlating microbial community compositions with environmental factors in activated sludge from four full-scale municipal wastewater treatment plants in shanghai, China applied microbiology and biotechnology. Appl. Microbiol. Biotechnol. 2016, 100, 4663–4673. [Google Scholar] [CrossRef]
- Xia, Y.; Wen, X.; Zhang, B.; Yang, Y. Diversity and assembly patterns of activated sludge microbial communities: A review. Biotechnol. Adv. 2018, 36, 1038–1047. [Google Scholar] [CrossRef]
- Han, I.; Yoo, K. Metagenomic profiles of antibiotic resistance genes in activated sludge, dewatered sludge and bioaerosols. Water 2020, 12, 1516. [Google Scholar] [CrossRef]
- Chen, H.J.; Lin, Y.Z.; Fanjiang, J.M.; Fan, C. Microbial community and treatment ability investigation in AOAO process for the optoelectronic wastewater treatment using PCR-DGGE biotechnology. Biodegradation 2013, 24, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Cydzik-Kwiatkowska, A.; Zielinska, M. Bacterial communities in full-scale wastewater treatment systems. World J. Microbiol. Biotechnol 2016, 32, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, X.-X.; Lu, X.; Liu, B.; Li, Y.; Long, C.; Li, A. Abundance and diversity of bacterial nitrifiers and denitrifiers and their functional genes in tannery wastewater treatment plants revealed by high-throughput sequencing. PLoS ONE 2014, 9, e113603. [Google Scholar] [CrossRef]
- Rodríguez, E.; García-Encina, P.A.; Stams, A.J.M.; Maphosa, F.; Sousa, D.Z. Meta-omics approaches to understand and improve wastewater treatment systems. Rev. Environ. Sci. Bio. Technol. 2015, 14, 385–406. [Google Scholar] [CrossRef]
- Nielsen, P.H.; Mielczarek, A.T.; Kragelund, C.; Nielsen, J.L.; Saunders, A.M.; Kong, Y.; Hansen, A.A.; Vollertsen, J. A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res. 2010, 44, 5070–5088. [Google Scholar] [CrossRef]
- Larsen, P.; Nielsen, J.L.; Otzen, D.; Nielsen, P.H. Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl. Environ. Microbiol. 2008, 74, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Bjornsson, L.; Hugenholtz, P.; Tyson, G.W.; Blackall, L.L. Filamentous chloroflexi (green non-sulfur bacteria) are abundant in wastewater treatment processes with biological nutrient removal. Microbiology 2002, 148, 2309–2318. [Google Scholar] [PubMed] [Green Version]
- Kragelund, C.; Caterina, L.; Borger, A.; Thelen, K.; Eikelboom, D.; Tandoi, V.; Kong, Y.; Van Der Waarde, J.; Krooneman, J.; Rossetti, S.; et al. Identity, abundance and ecophysiology of filamentous chloroflexi species present in activated sludge treatment plants. Fems. Microbiol. Ecol. 2007, 59, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hou, Y.; Ma, M.; Zhan, A. Potential pathogen communities in highly polluted river ecosystems: Geographical distribution and environmental influence. Ambio 2020, 49, 197–207. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grouping Program 1 | Grouping Program 2 | Sample | Temperature | pH | Conductivity | DO | SVI | Ammonium | Nitrite | Nitrate | TN | TP | COD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| °C | mg/L | ||||||||||||
| A | Stage 1 | BXAS1 | 28.40 | 7.44 | 203.90 | 3.18 | 12.50 | 25.26 | 0.08 | 0.27 | 26.78 | 2.03 | 53.47 |
| Stage 2 | BXAS2 | 29.00 | 7.07 | 540.40 | 0.63 | 35.36 | 16.83 | 0.08 | 1.30 | 17.84 | 1.83 | 37.47 | |
| BXAS3 | 29.50 | 6.81 | 490.70 | 0.72 | 39.60 | 7.34 | 0.17 | 3.26 | 12.74 | 1.30 | 44.13 | ||
| BXAS4 | 29.10 | 6.57 | 468.60 | 2.13 | 40.00 | 3.54 | 0.08 | 3.79 | 8.48 | 1.03 | 10.80 | ||
| Stage 3 | BXAS5 | 30.10 | 6.51 | 449.70 | 1.00 | 39.34 | 1.64 | 0.17 | 2.94 | 5.81 | 1.33 | 42.80 | |
| BXAS6 | 28.80 | 6.18 | 461.80 | 1.03 | 33.33 | 0.80 | 0.04 | 6.00 | 8.89 | 0.76 | 22.80 | ||
| B | Stage 1 | BXBS1 | 28.50 | 7.30 | 623.20 | 3.28 | 5.00 | 23.79 | 0.04 | 0.23 | 25.21 | 2.22 | 62.80 |
| Stage 2 | BXBS2.S | 31.70 | 6.82 | 547.30 | 0.16 | 33.00 | 6.92 | 0.12 | 0.74 | 9.07 | 1.29 | 32.13 | |
| BXBS3.S | 29.43 | 6.72 | 525.10 | 0.10 | 24.25 | 2.07 | 0.58 | 1.00 | 3.30 | 0.49 | 5.47 | ||
| BXBS4.S | 29.96 | 6.68 | 522.80 | 1.80 | 29.47 | 0.00 | 0.33 | 1.84 | 1.74 | 0.49 | 28.13 | ||
| BXBS2.N | 31.60 | 6.89 | 550.00 | 0.00 | 25.00 | 7.76 | 0.00 | 0.81 | 10.01 | 1.76 | 18.80 | ||
| BXBS3.N | 29.80 | 6.79 | 520.90 | 0.02 | 31.25 | 1.43 | 0.17 | 1.18 | 4.74 | 1.22 | 14.80 | ||
| BXBS4.N | 29.54 | 6.76 | 515.90 | 2.28 | 30.48 | 3.12 | 0.00 | 1.41 | 3.15 | 0.95 | 12.13 | ||
| Stage 3 | BXBS5 | 29.00 | 6.85 | 524.50 | 2.04 | 28.00 | 4.18 | 0.08 | 0.87 | 5.90 | 1.08 | 22.80 |
| Shannon | Chao 1 | Observed Species | PD Whole Tree | OTU Number | |
|---|---|---|---|---|---|
| BXAS1 | 7.8696 | 1782.69 | 1746 | 100.9319 | 1965 |
| BXAS2 | 8.4169 | 2155.26 | 1943 | 114.8954 | 2227 |
| BXAS3 | 8.8675 | 2264.85 | 2025 | 117.9512 | 2281 |
| BXAS4 | 8.6528 | 2145.23 | 1877 | 111.7669 | 2143 |
| BXAS5 | 7.5448 | 1600.30 | 1253 | 80.8964 | 1464 |
| BXAS6 | 7.6178 | 1591.58 | 1402 | 77.656 | 1574 |
| BXBS1 | 7.1054 | 1872.23 | 1640 | 99.3052 | 1926 |
| BXBS2.S | 7.5643 | 2051.50 | 1496 | 98.4937 | 1758 |
| BXBS3.S | 8.262 | 1911.95 | 1591 | 103.3599 | 1751 |
| BXBS4.S | 8.8196 | 2274.63 | 2045 | 118.6655 | 2360 |
| BXBS2.N | 8.7594 | 2130.87 | 1893 | 117.5945 | 2126 |
| BXBS3.N | 7.4933 | 1640.02 | 1245 | 86.3666 | 1469 |
| BXBS4.N | 8.7389 | 2192.48 | 1944 | 122.5322 | 2217 |
| BXBS5 | 9.0738 | 2818.80 | 2399 | 122.2691 | 2689 |
| Group | R-Value | p-Value |
|---|---|---|
| A–B | 0.1696 | 0.082 |
| Stage 1–Stage 3 | 0.8333 | 0.2 |
| Stage 2–Stage 3 | 0.2934 | 0.059 |
| Stage 1–Stage 2 | 0.7718 | 0.02 |
| r2 | Pr (>r) | |
|---|---|---|
| Temperature | 0.1682 | 0.3668 |
| pH | 0.3870 | 0.0634 |
| Conductivity | 0.0710 | 0.6866 |
| DO | 0.4238 | 0.0559 |
| SVI | 0.0880 | 0.6236 |
| Ammonium | 0.4049 | 0.0554 |
| Nitrite | 0.3004 | 0.1319 |
| Nitrate | 0.4276 | 0.0409 |
| TN | 0.4751 | 0.0294 |
| TP | 0.3490 | 0.0864 |
| COD | 0.4614 | 0.0374 |
| Physicochemical factors | 0.2873 | 0.1098 |
| Nutrients | 0.4500 | 0.0119 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, N.; Zhong, L.; Ouyang, L.; Xu, W.; Zeng, Q.; Wang, K.; Zaynab, M.; Chen, H.; Xu, F.; Li, S. Community Composition and Function of Bacteria in Activated Sludge of Municipal Wastewater Treatment Plants. Water 2021, 13, 852. https://doi.org/10.3390/w13060852
Xie N, Zhong L, Ouyang L, Xu W, Zeng Q, Wang K, Zaynab M, Chen H, Xu F, Li S. Community Composition and Function of Bacteria in Activated Sludge of Municipal Wastewater Treatment Plants. Water. 2021; 13(6):852. https://doi.org/10.3390/w13060852
Chicago/Turabian StyleXie, Ning, Liping Zhong, Liao Ouyang, Wang Xu, Qinghuai Zeng, Keju Wang, Madiha Zaynab, Huirong Chen, Fangfang Xu, and Shuangfei Li. 2021. "Community Composition and Function of Bacteria in Activated Sludge of Municipal Wastewater Treatment Plants" Water 13, no. 6: 852. https://doi.org/10.3390/w13060852