
Erythromycin Abatement from Water by Electro-Fenton and Peroxyelectrocoagulation Treatments

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instruments and Analytical Procedures
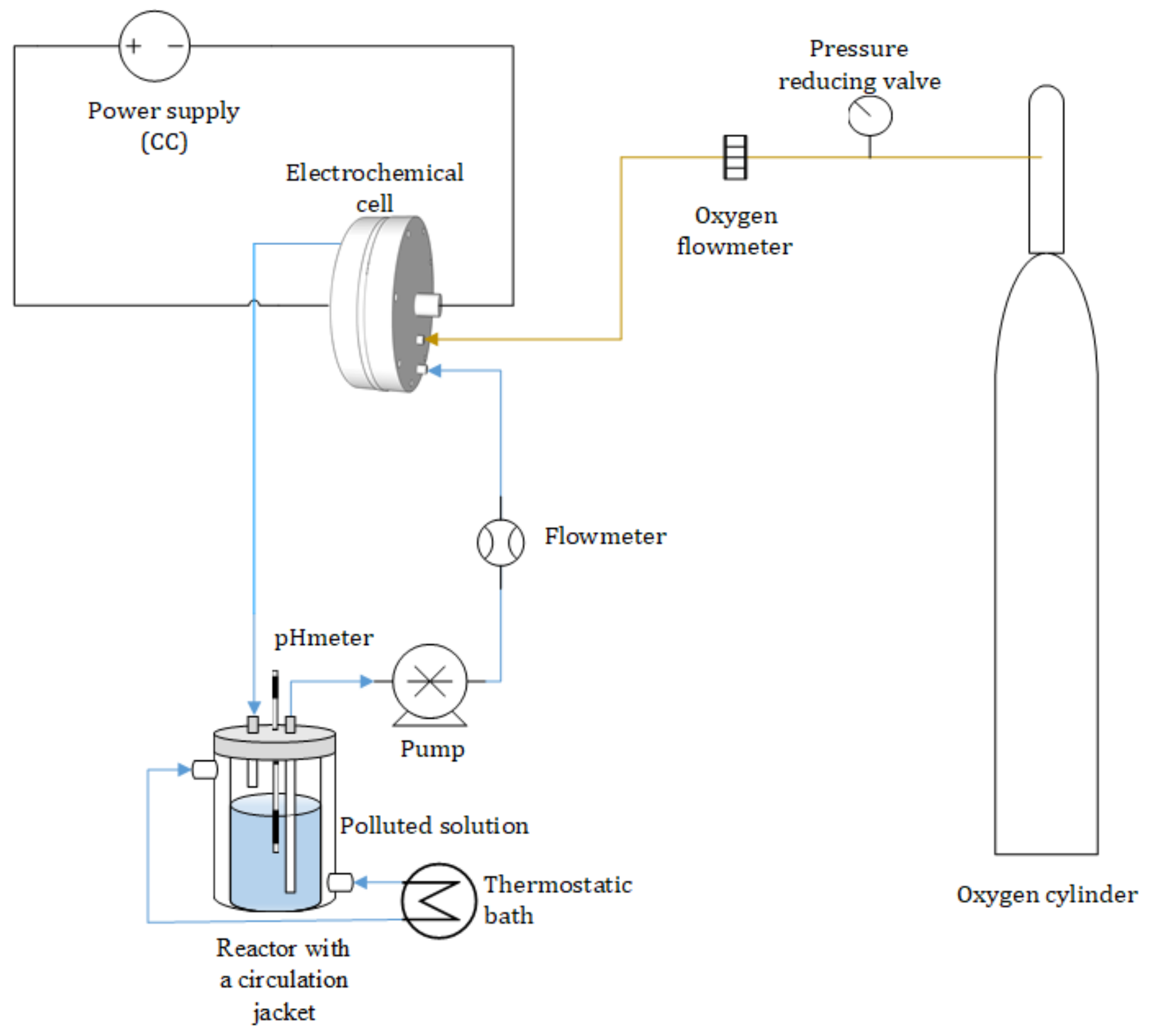
2.3. Experimental Setup and Operational Conditions
3. Results and Discussion
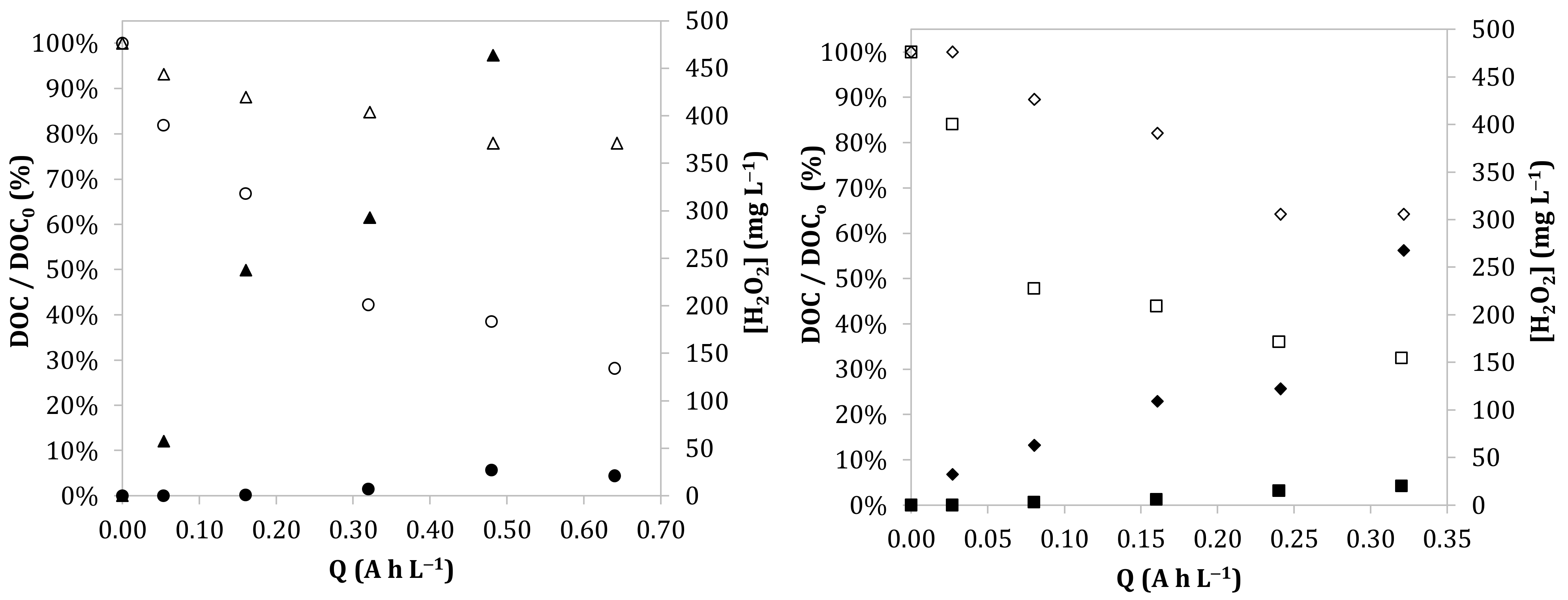
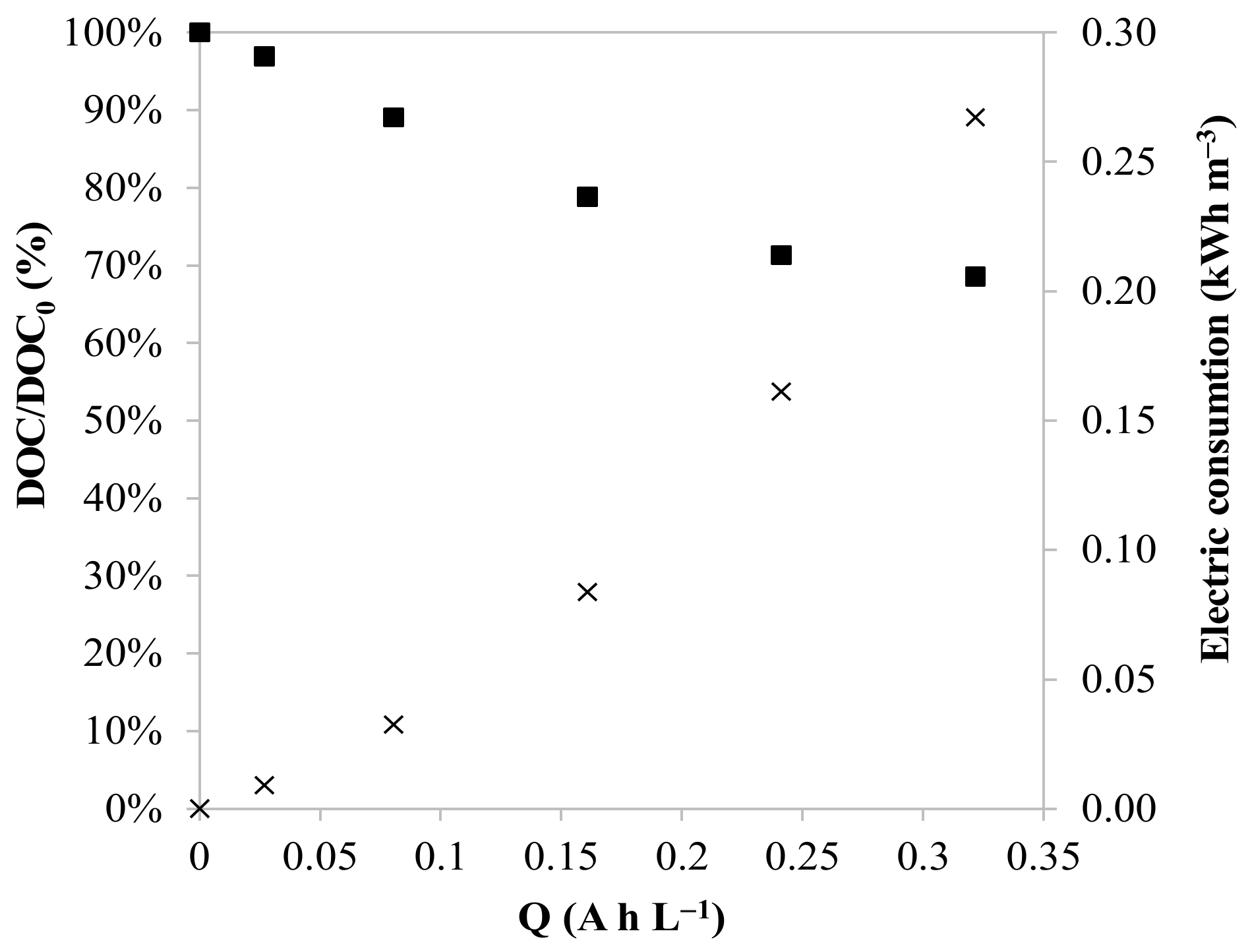
3.1. Effect of the Anode Material and the Current Density
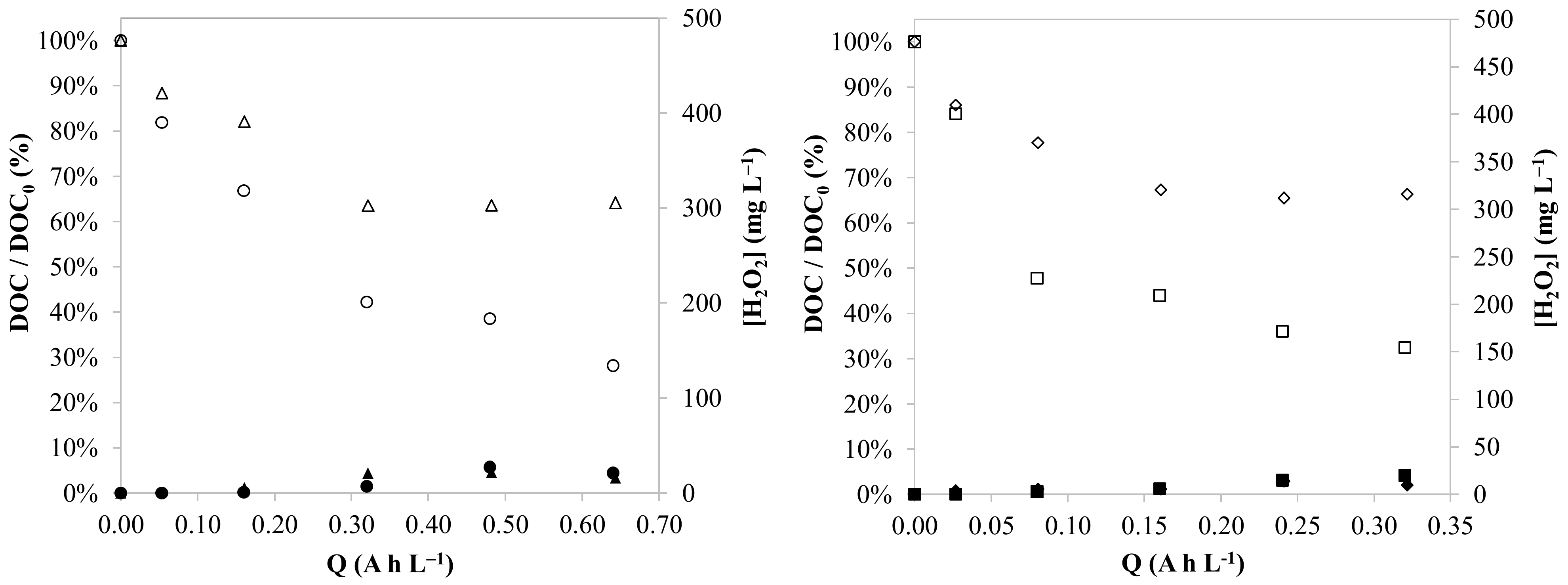
3.2. Effect of the Oxygen Flowrate
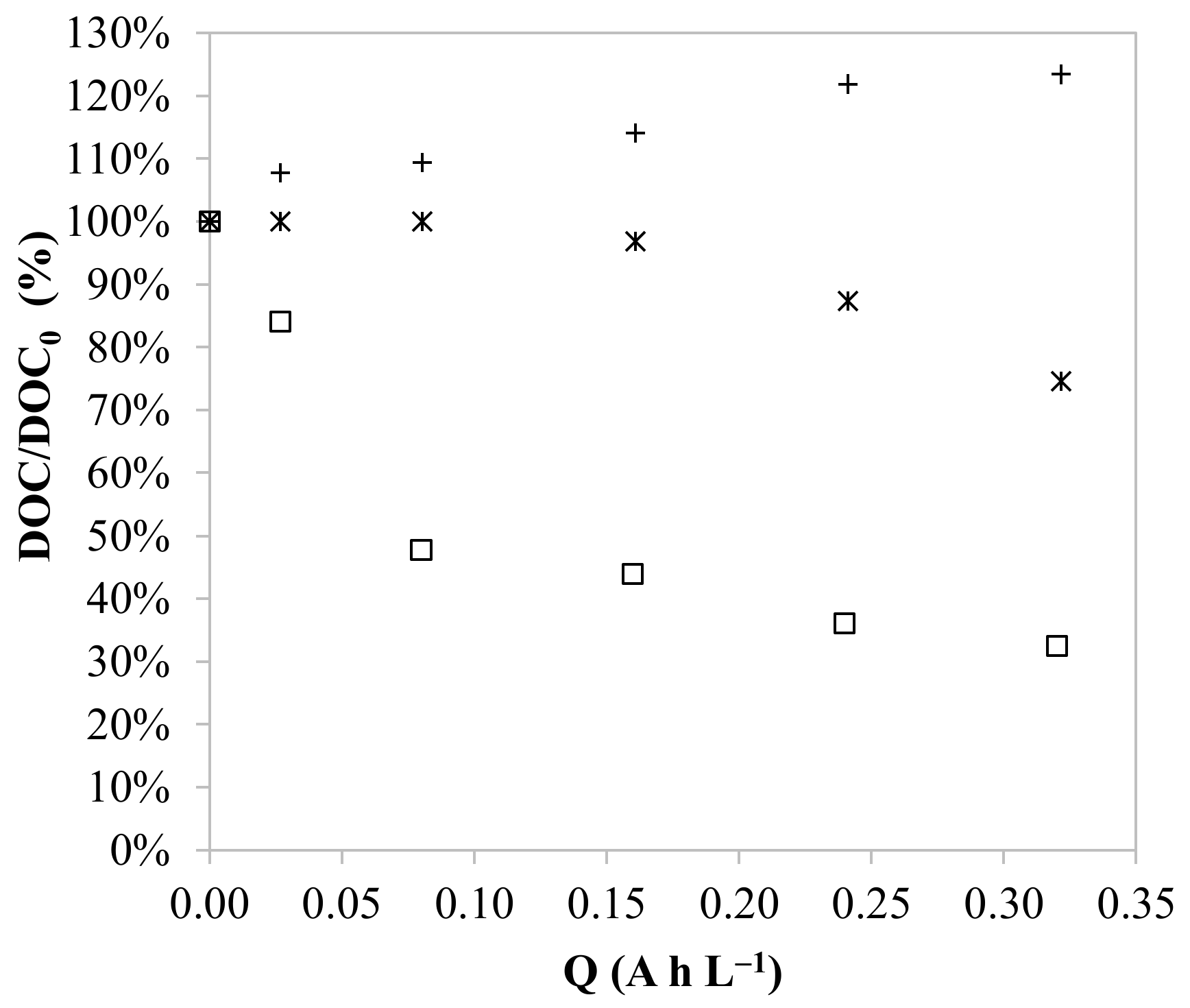
3.3. Effect of the pH
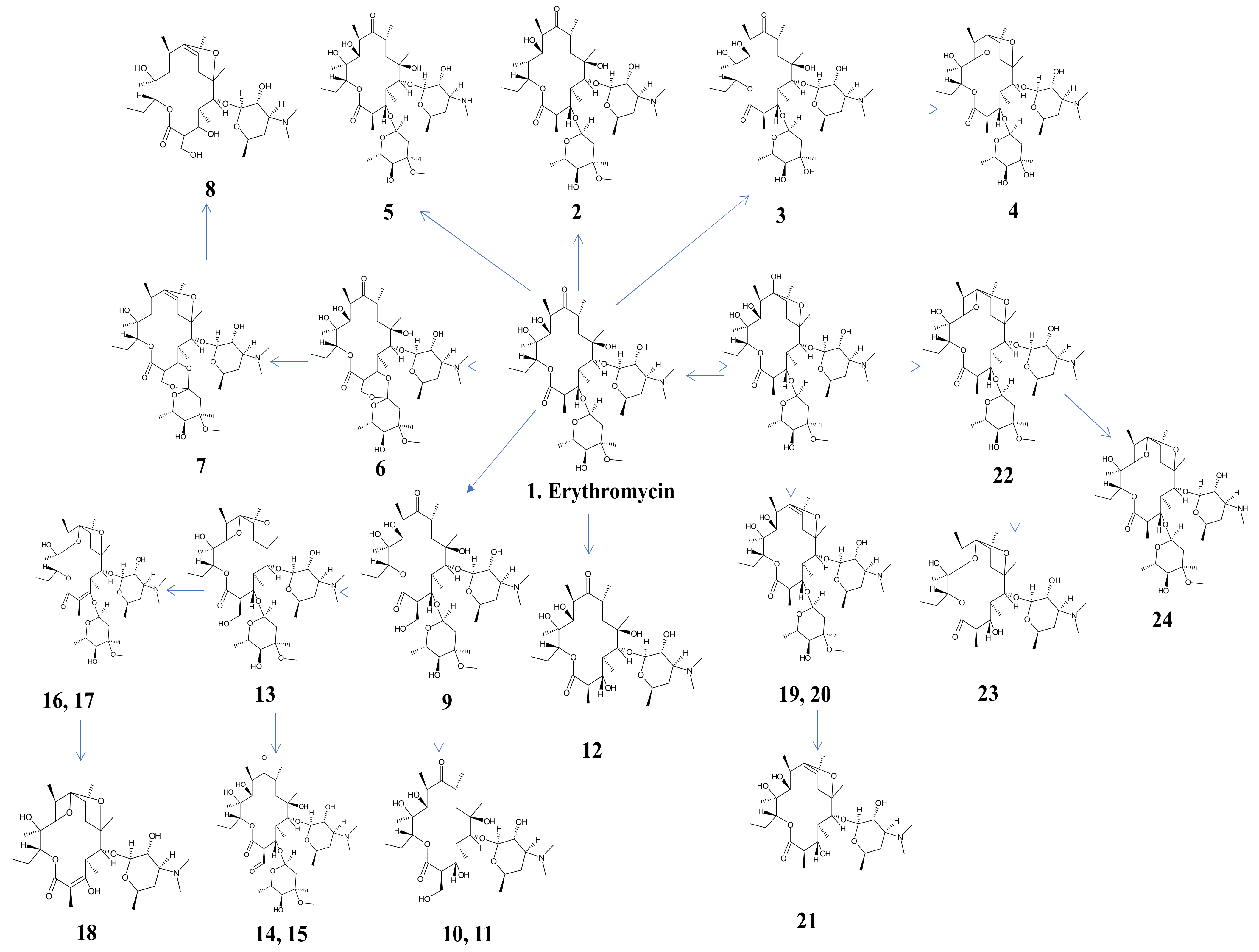
3.4. Identification of Aromatic Byproducts of Erythromycin and Degradation Pathway Proposed
3.5. Extrapolation of the PEC Process to a Real Effluent
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlsson, C.; Johansson, A.K.; Alvan, G.; Bergman, K.; Kühler, T. Are pharmaceuticals potent environmental pollutants? Part I: Environmental risk assessments of selected active pharmaceutical ingredients. Sci. Total Environ. 2006, 364, 67–87. [Google Scholar] [CrossRef]
- Sousa, J.C.G.; Ribeiro, A.R.; Barbosa, M.O.; Ribeiro, C.; Tiritan, M.E.; Pereira, M.F.R.; Silva, A.M.T. Monitoring of the 17 EU Watch List contaminants of emerging concern in the Ave and the Sousa Rivers. Sci. Total Environ. 2019, 649, 1083–1095. [Google Scholar] [CrossRef]
- Cunningham, V.L.; Binks, S.P.; Olson, M.J. Human health risk assessment from the presence of human pharmaceuticals in the aquatic environment. Regul. Toxicol. Pharmacol. 2009, 53, 39–45. [Google Scholar] [CrossRef]
- European Commission. Commission Implementing Decision (EU) 2015/495 of 20 March 2015 Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council; Office for Official Publications of the European Communities: Luxembourg, 2015; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32015D0495&from=EN (accessed on 18 April 2021).
- European Commission. Commission Implementing Decision (EU) 2018/840 of 5 June 2018 Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council and Repealing Commission Implementing Decision (EU) 2015/495; Office for Official Publications of the European Communities: Luxembourg, 2018; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018D0840&from=EN (accessed on 18 April 2021).
- European Commission. Commission Implementing Decision (EU) 2020/1161 of 4 August 2020 Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council; Office for Official Publications of the European Communities: Luxembourg, 2020; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020D1161&from=EN (accessed on 18 April 2021).
- Rodriguez-Mozaz, S.; Vaz-Moreira, I.; Varela Della Giustina, S.; Llorca, M.; Barceló, D.; Schubert, S.; Berendonk, T.U.; Michael-Kordatou, I.; Fatta-Kassinos, D.; Martinez, J.L.; et al. Antibiotic residues in final effluents of European wastewater treatment plants and their impact on the aquatic environment. Environ. Int. 2020, 140, 105733. [Google Scholar] [CrossRef]
- Polianciuc, S.I.; Gurzău, A.E.; Kiss, B.; Georgia Ștefan, M.; Loghin, F. Antibiotics in the environment: Causes and consequences. Med. Pharm. Rep. 2020, 93, 231–240. [Google Scholar] [CrossRef]
- Hanna, N.; Sun, P.; Sun, Q.; Li, X.; Yang, X.; Ji, X.; Zou, H.; Ottoson, J.; Nilsson, L.E.; Berglund, B.; et al. Presence of antibiotic residues in various environmental compartments of Shandong province in eastern China: Its potential for resistance development and ecological and human risk. Environ. Int. 2018, 114, 131–142. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antibiotic Resistance. 21 July 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 18 April 2021).
- Veseli, A.; Mullallari, F.; Balidemaj, F.; Berisha, L.; Švorc, L.; Arbneshi, T. Electrochemical determination of erythromycin in drinking water resources by surface modified screen-printed carbon electrodes. Microchem. J. 2019, 148, 412–418. [Google Scholar] [CrossRef]
- Carraro, E.; Bonetta, S.; Bertino, C.; Lorenzi, E.; Bonetta, S.; Gilli, G. Hospital effluents management: Chemical, physical, microbiological risks and legislation in different countries. J. Environ. Manag. 2016, 168, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Kovalova, L.; Siegrist, H.; von Gunten, U.; Eugster, J.; Hagenbuch, M.; Wittmer, A.; Moser, R.; McArdell, C.S. Elimination of micropollutants during post-treatment of hospital wastewater with powdered activated carbon, ozone, and UV. Environ. Sci. Technol. 2013, 47, 7899–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsen, K.; Ternes, T.; Werner, G.; Wallner, U.; Löffler, D.; Ziebuhr, W.; Witte, W.; Hacker, J. Impact of antibiotics on conjugational resistance gene transfer in Staphylococcus aureus in sewage. Environ. Microbiol. 2003, 5, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Rueda-Márquez, J.J.; Palacios-Villarreal, C.; Manzano, M.; Blanco, E.; Ramírez del Solar, M.; Levchuk, I. Photocatalytic degradation of pharmaceutically active compounds (PhACs) in urban wastewater treatment plants effluents under controlled and natural solar irradiation using immobilized TiO2. Sol. Energy 2020, 208, 480–492. [Google Scholar] [CrossRef]
- Llorca, M.; Rodríguez-Mozaz, S.; Couillerot, O.; Panigoni, K.; de Gunzburg, J.; Bayer, B.; Czaja, R.; Barceló, D. Identification of new transformation products during enzymatic treatment of tetracycline and erythromycin antibiotics at laboratory scale by an on-line turbulent flow liquid-chromatography coupled to a high resolution mass spectrometer LTQ-Orbitrap. Chemosphere 2015, 119, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moslah, B.; Hapeshi, E.; Jrad, A.; Fatta-Kassinos, D.; Hedhili., A. Simultaneous Decontamination of Seven Residual Antibiotics in Secondary Treated Effluents by Solar Photo-Fenton and Solar TiO2 Catalytic Processes. In Recent Advances in Environmental Science from the Euro-Mediterranean and Surrounding Regions. EMCEI 2017. Advances in Science, Technology & Innovation (IEREK Interdisciplinary Series for Sustainable Development); Kallel, A., Ksibi, M., Ben Dhia, H., Khélifi, N., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Klavarioti, M.; Mantzavinos, D.; Kassinos, D. Removal of residual pharmaceuticals from aqueous systems by advanced oxidation processes. Environ. Int. 2009, 35, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Brillas, E.; Martínez-Huitle, C.A. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods. An updated review. Appl. Catal. B Environ. 2015, 166–167, 603–643. [Google Scholar] [CrossRef]
- Lima, V.B.; Goulart, L.A.; Rocha, R.S.; Steter, J.R.; Lanza, M.R.V. Degradation of antibiotic ciprofloxacin by different AOP systems using electrochemically generated hydrogen peroxide. Chemosphere 2020, 247, 125807. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Y.; Zhang, J.; Luo, L.; Yang, Y.; Huang, H.; Peng, H.; Tang, L.; Mu, Y. Insight into electro-Fenton and photo-Fenton for the degradation of antibiotics: Mechanism study and research gaps. Chem. Eng. J. 2018, 347, 379–397. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of tartaric acid in the presence of iron. J. Chem. Soc. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Peres Ribeiro, J.; Nunes, M.I. Recent trends and developments in Fenton processes for industrial wastewater treatment—A critical review. Environ. Res. 2021, 197, 110957. [Google Scholar] [CrossRef] [PubMed]
- Sychev, A.Y.; Isak, V.G. Iron compounds and the mechanisms of the homogeneous catalysis of the activation of O2 and H2O2 and of the oxidation of organic substrates. Russ. Chem. Rev. 1995, 64, 1105–1129. [Google Scholar] [CrossRef]
- Vasudevan, S. An efficient removal of phenol from water by peroxi-electrocoagulation processes. J. Water Process Eng. 2014, 2, 53–57. [Google Scholar] [CrossRef]
- Sandhwar, V.K.; Prasad, B. Comparison of electrocoagulation, peroxi-electrocoagulation and peroxi-coagulation processes for treatment of simulated purified terephthalic acid wastewater: Optimization, sludge and kinetic analysis. Korean J. Chem. Eng. 2018, 35, 909–921. [Google Scholar] [CrossRef]
- Farhadi, S.; Aminzadeh, B.; Torabian, A.; Khatibikamal, V.; Alizadeh Fard, M. Comparison of COD removal from pharmaceutical wastewater by electrocoagulation, photoelectrocoagulation, peroxi-electrocoagulation and peroxi-photoelectrocoagulation processes. J. Hazard. Mater. 2012, 219–220, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Marselli, B.; Garcia-Gomez, J.; Michaud, P.-A.; Rodrigo, M.A.; Comninellis, C. Electrogeneration of hydroxyl radical on boron-doped diamond electrodes. J. Environ. Sci. 2003, 150, D79–D83. [Google Scholar] [CrossRef]
- Oturan, N.; Oturan, M.A. Chapter 8—Electro-Fenton Process: Background, New Developments, and Applications. In Electrochemical Water and Wastewater Treatment; Martínez-Huitle, C.A., Rodrigo, M.A., Scialdone, O., Eds.; Butterworth-Heinemann: Oxford, UK, 2018; pp. 193–221. ISBN 9780128131602. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Ge, R.; Yang, S.; Zhai, Y.; Hua, T.; Ondon, B.S.; Zhou, Q.; Li, F. Microbial electro-Fenton: A promising system for antibiotics resistance genes degradation and energy generation. Sci. Total Environ. 2020, 699, 134160. [Google Scholar] [CrossRef] [PubMed]
- Pérez, T.; Sirés, I.; Brillas, E.; Nava, J.L. Solar photoelectro-Fenton flow plant modeling for the degradation of the antibiotic erythromycin in sulfate medium. Electrochim. Acta 2017, 228, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Sánchez Ruíz, C. Fenton Reactions (FS-TER-003). Inditex. 2015. Available online: https://www.wateractionplan.com/documents/177327/558166/Fenton+reactions.pdf/087c01a6-7f9c-2f33-95e7-b8a5945b9162 (accessed on 12 April 2021).
- APHA. Method 3500-Fe B. ASTMD 1068-77, Iron in Water, Test Method. In Standard Methods Standard Methods for the Examination of Water and Wastewater, 22nd ed.; American Public Health Association: Washington, DC, USA, 2012. [Google Scholar]
- Welcher, F.J. Standard Methods of Chemical Analysis, 6th ed.; Huntington 2B; R.E. Krieger Publishing Company: Florida, FL, USA, 1975; pp. 1827–1828. [Google Scholar]
- Hassanzadeh, A.; Barber, J.; Morris, G.A.; Gorry, P.A. Mechanism for the degradation of erythromycin A and erythromycin A 2′-Ethyl succinate in acidic aqueous solution. J. Phys. Chem. A 2007, 111, 10098–10104. [Google Scholar] [CrossRef]
- Volmer, D.A.; Hui, J.P. Study of erythromycin A decomposition products in aqueous solution by solid-phase microextraction/liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1998, 12, 123–129. [Google Scholar] [CrossRef]
- Chitneni, S.K.; Govaerts, C.; Adams, E.; Van Schepdael, A.; Hoogmartens, J. Identification of impurities in erythromycin by liquid chromatography–mass spectrometric detection. J. Chromatogr. A 2004, 1056, 111–120. [Google Scholar] [CrossRef]
- Deubel, A.; Fandino, A.S.; Sörgel, F.; Holzgrabe, U. Determination of erythromycin and related substances in commercial samples using liquid chromatography/ion trap mass spectrometry. J. Chromatogr. A 2006, 1136, 39–47. [Google Scholar] [CrossRef]
- Luiz, D.B.; Genena, A.K.; Virmond, E.; José, H.J.; Moreira, R.F.; Gebhardt, W.; Schröder, H.F. Identification of degradation products of erythromycin A arising from ozone and advanced oxidation process treatment. Water Environ. Res. 2010, 82, 797–805. [Google Scholar] [CrossRef]
- Malvar, J.L.; Abril, C.; Martín, J.; Santos, J.L.; Aparicio, I.; Escot, C.; Basanta, A.; Alonso, E. Development of an analytical method for the simultaneous determination of the 17 EU Watch List compounds in surface waters: A Spanish case study. Environ. Chem. 2018, 15, 493–505. [Google Scholar] [CrossRef]
- Ganzenko, O.; Trellu, C.; Oturan, N.; Huguenot, D.; Péchaud, Y.; van Hullebusch, E.D.; Oturan, M.A. Electro-Fenton treatment of a complex pharmaceutical mixture: Mineralization efficiency and biodegradability enhancement. Chemosphere 2020, 253, 126659. [Google Scholar] [CrossRef] [PubMed]
- Pignatello, J.J.; Liu, D.; Huston, P. Evidence for an additional oxidant in the photoassisted Fenton reaction. Environ. Sci. Technol. 1999, 33, 1832–1839. [Google Scholar] [CrossRef]
- Wu, K.; Xie, Y.; Zhao, J.; Hidaka, H. Photo-Fenton degradation of a dye under visible light irradiation. J. Mol. Catal. A Chem. 1999, 144, 77–84. [Google Scholar] [CrossRef]
- Diari Oficial de la Generalitat de Catalunya. DECRET 130/2003, de 13 de maig, pel qual s’aprova el Reglament dels serveis públics de sanejament. Diari Oficial de la Generalitat de Catalunya 2003, 3894, 11143–11158. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical-Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen-Atoms and Hydroxyl Radicals(•OH/•O–) in Aqueous-Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Grundl, T.; Delwiche, J. Kinetics of ferric oxyhydroxide precipitation. J. Contam. Hydrol. 1993, 4, 71–87. [Google Scholar] [CrossRef]
- Faust, B.C.; Hoigné, J. Photolysis of Fe(III)-hydroxy complexes as source of OH radical in clouds, fog and rain. Atmos. Environ. 1990, 24, 79–89. [Google Scholar] [CrossRef]
- Gallard, H.; De Laat, J.; Legube, B. Spectrophotometric study of the formation of iron(III)-hydroperoxy complexes in homogeneous aqueous solutions. Water Res. 1999, 33, 2929–2936. [Google Scholar] [CrossRef]
- Serra-Clusellas, A.; De Angelis, L.; Lin, C.-H.; Vo, P.; Bayati, M.; Sumner, L.; Lei, Z.; Amaral, N.B.; Bertini, L.M.; Mazza, J.; et al. Abatement of 2,4-D by H2O2 solar photolysis and solar photo-Fenton-like process with minute Fe(III) concentrations. Water Res. 2018, 144, 572–580. [Google Scholar] [CrossRef]
- Hirsch, R.; Ternes, T.; Haberer, K.; Kratz, K.-L. Occurrence of antibiotics in the aquatic environment. Sci. Total Environ. 1999, 225, 109–118. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, L.; Wang, B.; Liu, C.S.; Jia, Y.; Zhai, N.; Blaney, L.; Yu, G. Efficient multiresidue determination method for 168 pharmaceuticals and metabolites: Optimization and application to raw wastewater, wastewater effluent, and surface water in Beijing, China. Environ. Pollut. 2020, 261, 114113. [Google Scholar] [CrossRef] [PubMed]
- Eurostat. Electricity Prices (Including Taxes) for Household Consumers, First Half 2020. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php/Electricity_price_statistics (accessed on 5 March 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (min) | ESI | Quantitative Transition (m/z) | Qualitative Transition (m/z) | Collision Energy (V) | r2 | Quantification Limit (MLOQ) (ng L−1) | Detection Limit (MLOD) (ng L−1) |
|---|---|---|---|---|---|---|---|---|
| 17-β-Estradiol-d5 | 8.70 | ESI (−) | 276.2→147.1 | 276.2→187.2 | 49/41 | |||
| 17-β-Estradiol (E2) | 8.75 | ESI (−) | 271.2→145.1 | 271.2→183.1 | 45/49 | 0.99 | 1 | 0.3 |
| Estrone-d4 | 8.76 | ESI (−) | 273.1→147.1 | 273.1→145.0 | 49/60 | |||
| Estrone (E1) | 8.79 | ESI (−) | 269.1→145.1 | 269.1→143.1 | 41/60 | 0.99 | 1 | 0.3 |
| 17-α-Ethinylestradiol-d5 | 8.90 | ESI (−) | 299.2→147.1 | 299.2→269.1 | 33/53 | |||
| 17-α-Ethinylestradiol (EE2) | 8.85 | ESI (−) | 295.2→145.2 | 295.17→142.9 | 49/60 | 0.99 | 1 | 0.03 |
| Amoxicillin-d4 | 2.71 | ESI (+) | 370.1→114.0 | 370.1→353.3 | 21/9 | |||
| Amoxicillin | 2.73 | ESI (+) | 366.1→114.0 | 366.1→349.2 | 18/4 | 0.95 | 1 | 0.3 |
| Thiamethoxam-d4 | 4.78 | ESI (+) | 296.1→215.0 | 296.1→183.0 | 5/21 | |||
| Thiamethoxam | 4.82 | ESI (+) | 292.0→210.9 | 292.0→180.8 | 9/21 | 0.99 | 1 | 0.3 |
| Ciprofloxacin | 5.39 | ESI (+) | 332.1→231.0 | 332.0→314.2 | 45/17 | 0.99 | 1 | 0.3 |
| Clothianidin-d3 | 4.47 | ESI (+) | 253.0→172.0 | 253.0→131.8 | 9/21 | |||
| Clothianidin | 4.48 | ESI (+) | 250.0→131.9 | 250.0→169.1 | 13/9 | 0.99 | 1 | 0.3 |
| Imidacloprid-d4 | 5.49 | ESI (+) | 260.1→179.2 | 260.1→213.1 | 13/9 | |||
| Imidacloprid | 5.52 | ESI (+) | 256.1→175.0 | 256.1→209.1 | 1721 | 0.99 | 10 | 3 |
| Acetamiprid-d3 | 5.94 | ESI (+) | 226.1→126.1 | 226.1→59.2 | 21/13 | |||
| Acetamiprid | 5.95 | ESI (+) | 223.1→126.1 | 223.1→56.0 | 17/13 | 0.99 | 1 | 0.3 |
| Thiacloprid | 6.42 | ESI (+) | 253.0→125.9 | 253.0→98.9 | 21/53 | 0.99 | 1 | 0.3 |
| Azithromycin | 7.25 | ESI (+) | 749.5→82.9 | 749.5→591.3 | 60/33 | 0.99 | 1 | 0.3 |
| Erythromycin-d3 | 9.12 | ESI (+) | 737.5→161.0 | 737.5→82.9 | 37/60 | |||
| Erythromycin | 9.12 | ESI (+) | 734.4→158.1 | 734.4→83.1 | 37/60 | 0.99 | 6 | 1.8 |
| Methiocarb-d3 | 10.36 | ESI (+) | 229.1→169.1 | 229.1→121.0 | 9/17 | |||
| Methiocarb | 10.38 | ESI (+) | 226.1→169.0 | 226.1→121.1 | 17/9 | 0.99 | 20 | 6 |
| Clarithromycin-d3 | 10.84 | ESI (+) | 751.5→161.1 | 751.5→83.1 | 25/57 | |||
| Clarithromycin | 10.86 | ESI (+) | 748.5→158.2 | 748.5→83.1 | 37/60 | 0.99 | 1 | 0.3 |
| Metaflumizone | 14.60 | ESI (+) | 507.1→178.2 | 507.1→87.0 | 33/25 | 0.99 | 6 | 1.8 |
| Experiment Conditions | Type of Water | Anode Type | janode (mA/cm2) | Oxygen Flowrate (L/min) | pH |
|---|---|---|---|---|---|
| EF-1 | Ultrapure water. 10 mg L−1 erythromycin and 0.05 M Na2SO4 | BDD | 5 | 2.0 | 2.8 |
| EF-2 | BDD | 10 | 2.0 | 2.8 | |
| PEC-1 | Fe | 5 | 2.0 | 2.8 | |
| PEC-2 | Fe | 10 | 2.0 | 2.8 | |
| PEC-3 | Fe | 5 | 0.8 | 2.8 | |
| PEC-4 | Fe | 10 | 0.8 | 2.8 | |
| PEC-5 | Fe | 5 | 2.0 | 5.0 | |
| PEC-6 | Fe | 5 | 2.0 | 7.0 | |
| PEC-7 | Effluent from a tertiary treatment of an urban WWTP | Fe | 5 | 2.0 | 2.8 |
| N° | Compound | Molecular Formula | Pseudo-Molecular Ion (m/z) | Retention Time (min) | Compound Concentration (µg L−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 min pH 2.8 | 5 min | 15 min | 30 min | 45 min | 60 min | |||||
| 1 | Erythromycin (Erythromycin A) | C37H67NO13 | 734.4685 | 4.0 | 457.4 | 17.9 | 0.8 | Non detected (n.d.) | n.d. | n.d. |
| 2 | Erythromycin B | C37H67NO12 | 718.4736 | 4.6 | 204.1 | 47.0 | 0.5 | n.d. | n.d. | n.d. |
| 3 | Erythromycin C | C36H65NO13 | 720.4529 | 3.5 | 5.2 | n.d. | n.d. | n.d. | n.d. | n.d. |
| 4 | Erythromycin C anhydrous | C36H63NO12 | 702.4423 | 4.2 | 61.1 | 19.2 | 0.2 | n.d. | n.d. | n.d. |
| 5 | Erythromycin A desosamine N-demethylation | C36H65NO13 | 720.4529 | 3.9 | 13.5 | 2.3 | 1.5 | n.d. | n.d. | n.d. |
| 6 | Erythromycin E | C37H65NO14 | 748.4478 | 3.9 | 301.3 | 71.0 | 2.9 | n.d. | n.d. | n.d. |
| 7 | Erythromycin E dehydration + dehydroxylation | C37H63NO12 | 714.4423 | 5.2 | 22.1 | 99.3 | 1.2 | n.d. | n.d. | n.d. |
| 8 | Erythromycin E dehydration + dehydroxylation—cladinose | C29H49NO9 | 556.348 | 4.3 | n.d. | 46.7 | 5.0 | n.d. | n.d. | n.d. |
| 9 | Erythromycin F | C37H67NO14 | 750.4634 | 3.7 | 241.7 | 14.5 | n.d. | n.d. | n.d. | n.d. |
| 10 | Erythromycin F—cladinose TP-1 | C29H53NO11 | 592.3691 | 2.5 | 99.1 | 43.6 | 2.3 | n.d. | n.d. | n.d. |
| 11 | Erythromycin F—cladinose TP-2 | C29H53NO11 | 592.3691 | 2.6 | 1.2 | 64.3 | 3.2 | n.d. | n.d. | n.d. |
| 12 | Erythromycin A—cladinose | C29H53NO10 | 576.3742 | 3.0 | 68.1 | 149.0 | 9.1 | n.d. | n.d. | n.d. |
| 13 | Erythromycin F enol ether or anhydrous | C37H65NO13 | 732.4529 | 3.6 | 3.5 | 20.8 | 0.8 | n.d. | n.d. | n.d. |
| 14 | Erythromycin F dehydrogenation TP-1 | C37H65NO14 | 748.4478 | 3.5 | 0.7 | 31.4 | 0.8 | n.d. | n.d. | n.d. |
| 15 | Erythromycin F dehydrogenation TP-2 | C37H65NO14 | 748.4478 | 3.9 | 12.8 | 61.0 | 3.1 | n.d. | n.d. | n.d. |
| 16 | Erythromycin F enol ether or anhydrous + lactone dehydration TP-1 | C37H63NO12 | 714.4423 | 4.4 | 40.6 | 21.1 | 0.2 | n.d. | n.d. | n.d. |
| 17 | Erythromycin F enol ether or anhydrous + lactone dehydration TP-2 | C37H63NO12 | 714.4423 | 4.8 | 20.9 | 27.4 | 0.2 | n.d. | n.d. | n.d. |
| 18 | Erythromycin F enol ether or anhydrous + lactone dehydration—cladinose | C29H49NO9 | 556.348 | 3.0 | n.d. | 5.6 | 0.4 | n.d. | n.d. | n.d. |
| 19 | Erythromycin A enol ether TP-1 | C37H65NO12 | 716.458 | 4.8 | 5.1 | 4.4 | n.d. | n.d. | n.d. | n.d. |
| 20 | Erythromycin A enol ether TP-2 | C37H65NO12 | 716.458 | 5.2 | 1.8 | 8.6 | 0.1 | n.d. | n.d. | n.d. |
| 21 | Erythromycin A enol ether—cladinose | C29H51NO9 | 558.3637 | 4.2 | 20.5 | 7.2 | n.d. | n.d. | n.d. | n.d. |
| 22 | Erythromycin A anhydrous | C37H65NO12 | 716.458 | 4.5 | 1533.5 | 432.0 | 2.6 | n.d. | n.d. | n.d. |
| 23 | Erythromycin A anhydrous desosamine N-demethylation | C36H63NO12 | 702.4423 | 4.4 | 56.2 | 3.5 | n.d. | n.d. | n.d. | n.d. |
| 24 | Erythromycin A anhydrous—cladinose | C29H51NO9 | 558.3637 | 3.5 | 46.7 | 79.4 | 4.7 | n.d. | n.d. | n.d. |
| Parameter | Units | Value |
|---|---|---|
| pH | - | 7.6 |
| Electrical conductivity | ms cm−1 | 1.5 |
| Total Inorganic Carbon | mg L−1 | 72.8 |
| DOC | mg L−1 | 9.9 |
| Total N | mg L−1 | 33.1 |
| Compound | Effluent from the Tertiary Treatment of an Urban WWTP (ng L−1) | Treated Effluent from the PEC Treatment (ng L−1) | Removals (%) |
|---|---|---|---|
| E1 | 858 | 5 | 99% |
| E2 | 127 | <MLOQ | >99% |
| EE2 | 117 | 1 | 99% |
| Amoxicillin trihydrate | <MLOQ | <MLOQ | - |
| Thiamethoxam | 7 | <MLOQ | >86% |
| Ciprofloxacin | 1 | <MLOQ | - |
| Clothianidin | 6 | <MLOQ | >83% |
| Imidacloprid | 60 | <MLOQ | >83% |
| Acetamiprid | 30 | <MLOQ | >97% |
| Thiacloprid | 21 | <MLOQ | >95% |
| Azythromycin | 365 | <MLOQ | 100% |
| Erythromycin | 23 | <MLOQ | >74% |
| Methiocarb | 30 | <MLOQ | >33% |
| Clarythromycin | 31 | <MLOQ | >97% |
| Metaflumizone | 17 | <MLOQ | >65% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra-Clusellas, A.; Sbardella, L.; Herrero, P.; Delpino-Rius, A.; Riu, M.; de Lourdes Correa, M.; Casadellà, A.; Canela, N.; Martínez-Lladó, X. Erythromycin Abatement from Water by Electro-Fenton and Peroxyelectrocoagulation Treatments. Water 2021, 13, 1129. https://doi.org/10.3390/w13081129
Serra-Clusellas A, Sbardella L, Herrero P, Delpino-Rius A, Riu M, de Lourdes Correa M, Casadellà A, Canela N, Martínez-Lladó X. Erythromycin Abatement from Water by Electro-Fenton and Peroxyelectrocoagulation Treatments. Water. 2021; 13(8):1129. https://doi.org/10.3390/w13081129
Chicago/Turabian StyleSerra-Clusellas, Anna, Luca Sbardella, Pol Herrero, Antoni Delpino-Rius, Marc Riu, María de Lourdes Correa, Anna Casadellà, Núria Canela, and Xavier Martínez-Lladó. 2021. "Erythromycin Abatement from Water by Electro-Fenton and Peroxyelectrocoagulation Treatments" Water 13, no. 8: 1129. https://doi.org/10.3390/w13081129