
ALKBH4 Stabilization Is Required for Arsenic-Induced 6mA DNA Methylation Inhibition, Keratinocyte Malignant Transformation, and Tumorigenicity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid and Lentivirus Generation and Infection
2.3. Quantitative Real-Time PCR (qPCR)
2.4. The 6mA Dot Blot Assay
2.5. Quantification of m6A, m1A, and Am Levels in mRNA by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry (UHPLC-MS/MS) Assay
2.6. Immunoblotting
2.7. Soft Agar Colony Formation and Cell Proliferation Assay
2.8. Tumorigenicity Assay in Immunocompromised Mice
2.9. Statistical Analyses
3. Results
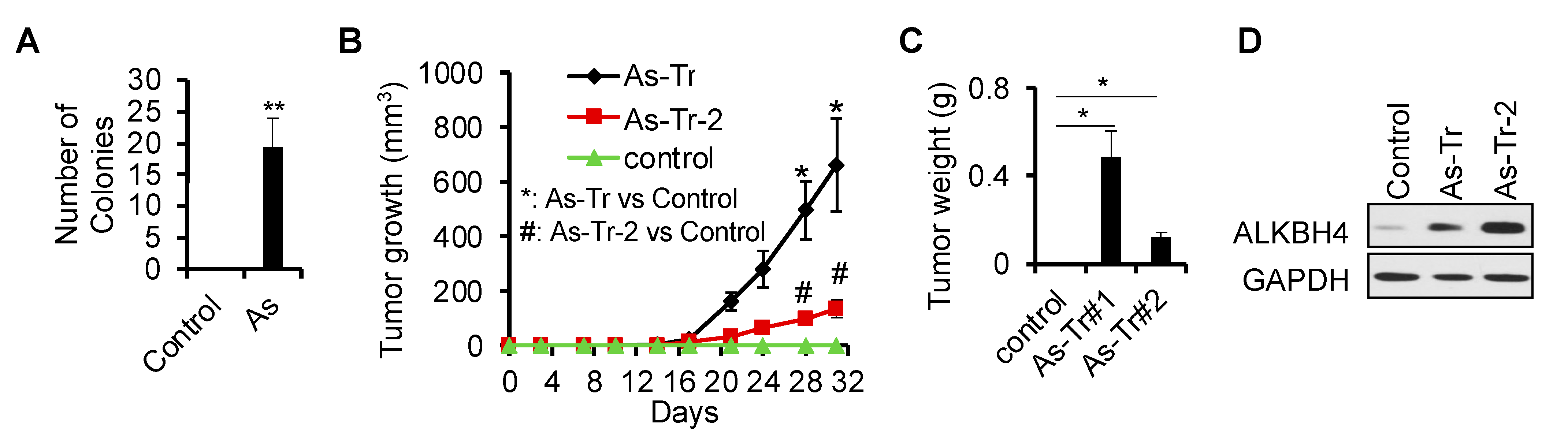
3.1. ALKBH4 Upregulated in Arsenic-Induced Skin Cancer
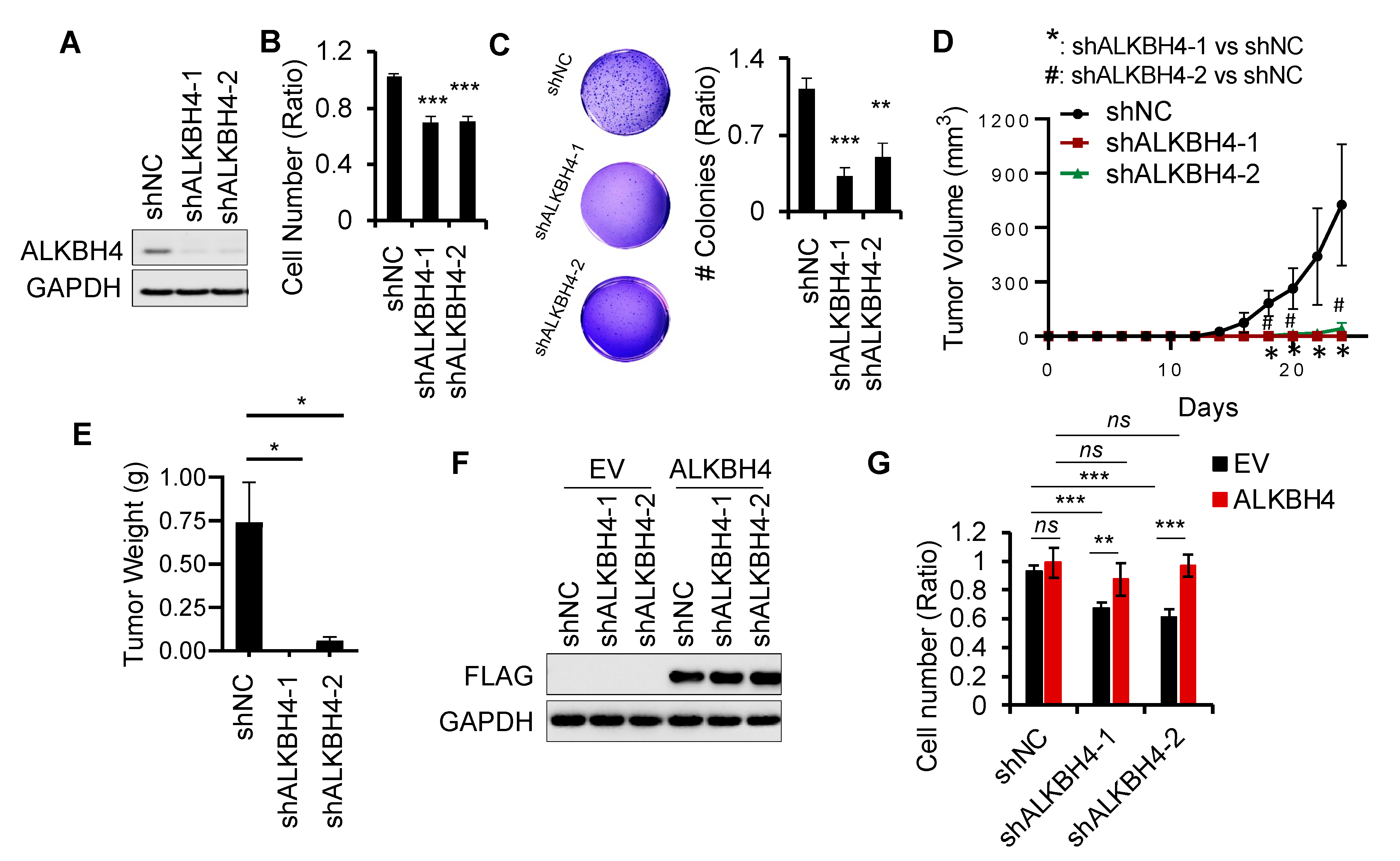
3.2. ALKBH4 Is Required for Arsenic-Induced Tumorigenicity
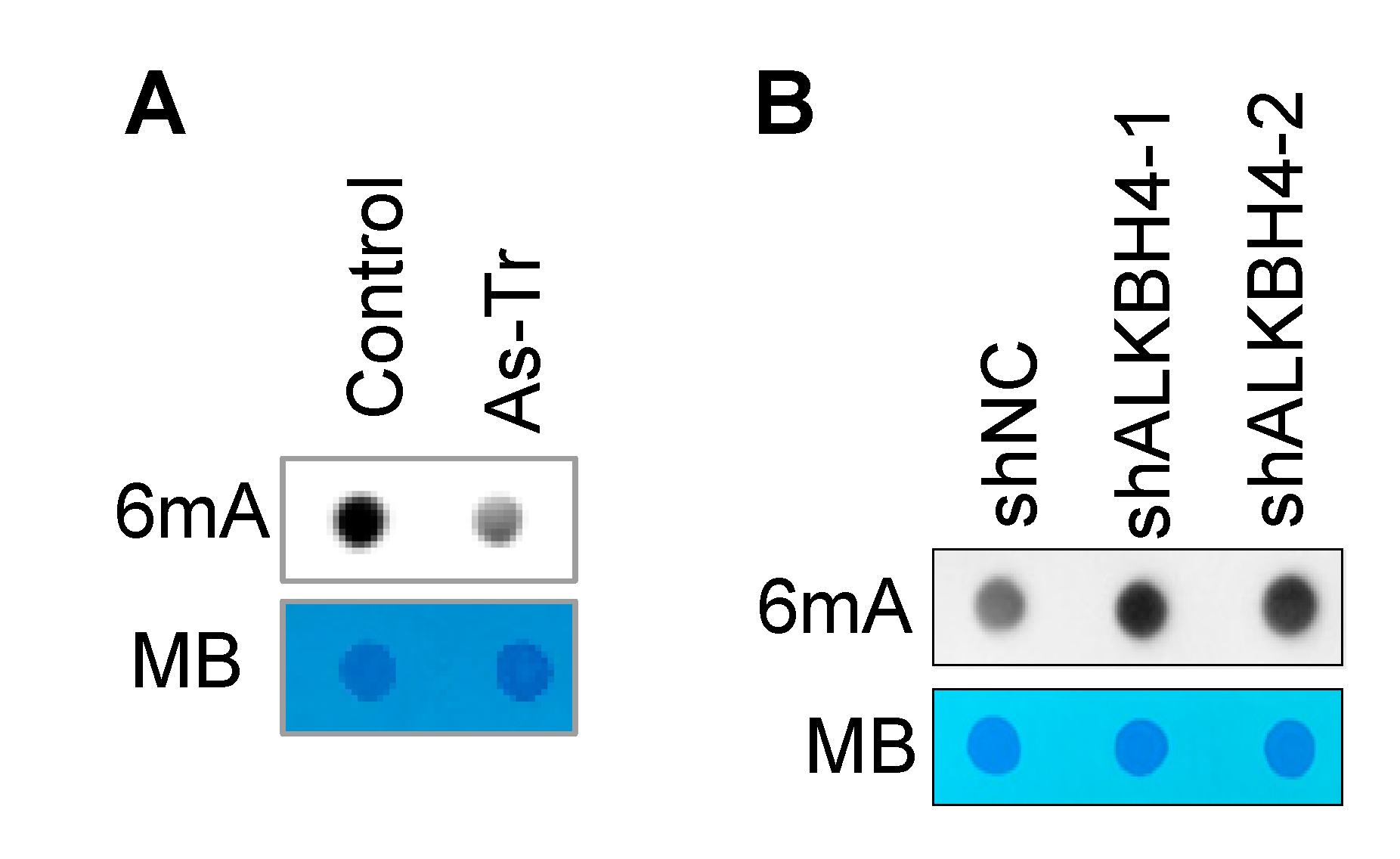
3.3. ALKBH4 Upregulation Inhibits DNA 6mA Methylation in Arsenic-Transformed Cells
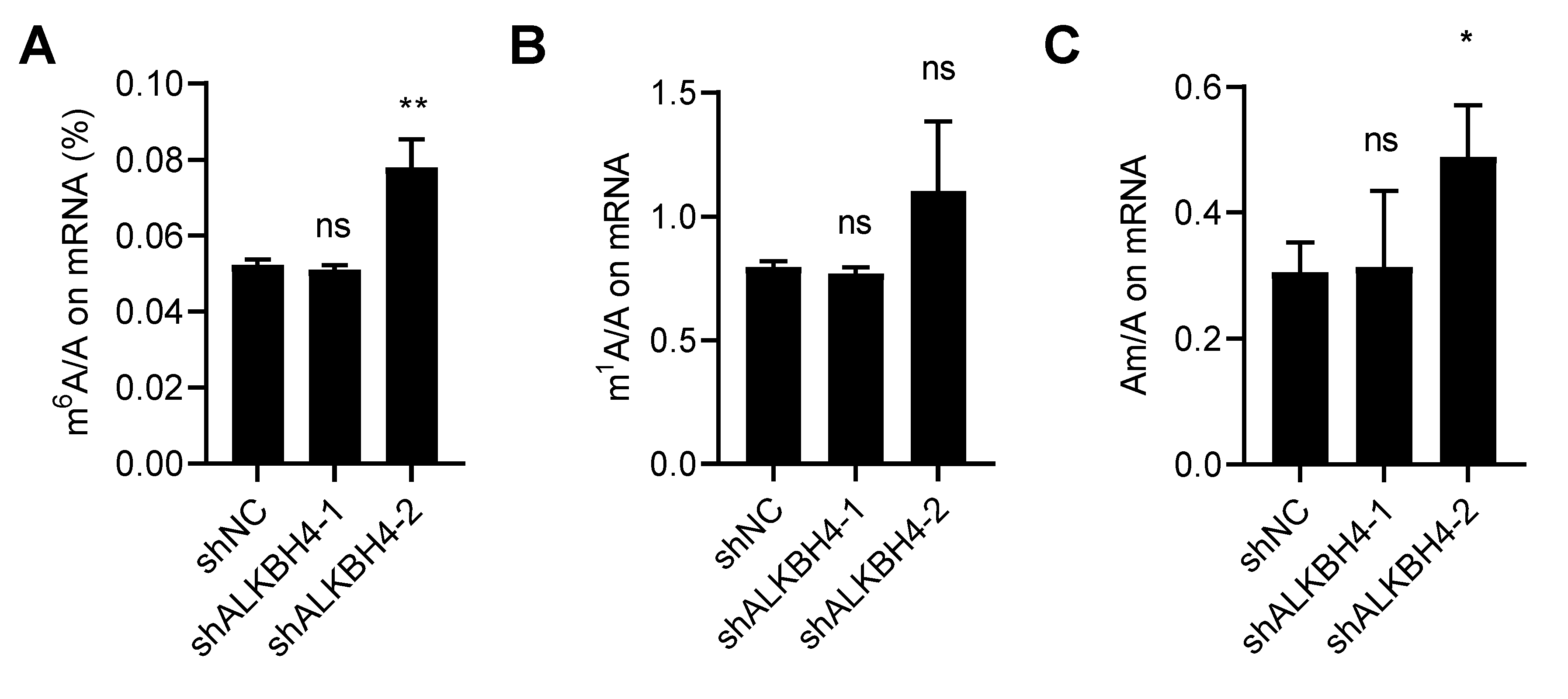
3.4. ALKBH4 Does Not Regulate mRNA Modification of Arsenic Treatment Response
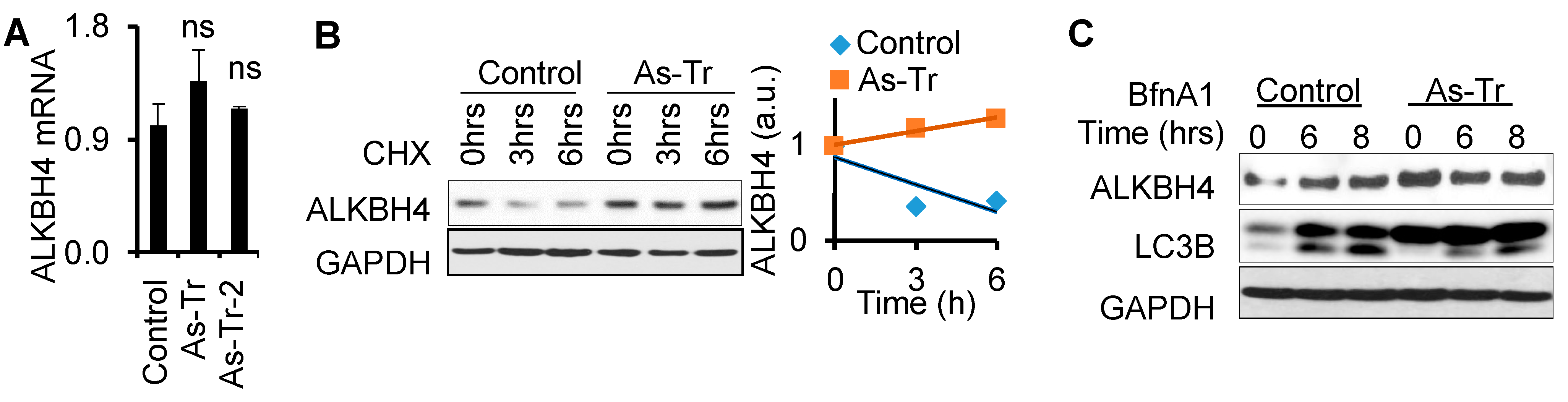
3.5. ALKBH4 Protein Stability Is Upregulated by Arsenic via Autophagy Inhibition
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nordstrom, D.K. Public health. Worldwide occurrences of arsenic in ground water. Science 2002, 296, 2143–2145. [Google Scholar] [CrossRef]
- Chung, J.Y.; Yu, S.D.; Hong, Y.S. Environmental source of arsenic exposure. J. Prev. Med. Public Health 2014, 47, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Mandal, B.K.; Suzuki, K.T. Arsenic round the world: A review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Shankar, S.; Shanker, U.; Shikha. Arsenic contamination of groundwater: A review of sources, prevalence, health risks, and strategies for mitigation. Sci. World J. 2014, 2014, 304524. [Google Scholar] [CrossRef]
- Torchia, D.; Massi, D.; Caproni, M.; Fabbri, P. Multiple cutaneous precanceroses and carcinomas from combined iatrogenic/professional exposure to arsenic. Int. J. Dermatol. 2008, 47, 592–593. [Google Scholar] [CrossRef]
- Matthews, N.H.; Fitch, K.; Li, W.Q.; Morris, J.S.; Christiani, D.C.; Qureshi, A.A.; Cho, E. Exposure to Trace Elements and Risk of Skin Cancer: A Systematic Review of Epidemiologic Studies. Cancer Epidemiol. Biomark. Prev. 2019, 28, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Diwan, B.A.; Sun, Y.; Liu, J.; Qu, W.; He, Y.; Styblo, M.; Waalkes, M.P. Arsenic-induced malignant transformation of human keratinocytes: Involvement of Nrf2. Free Radic. Biol. Med. 2008, 45, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Graziano, J.H.; Parvez, F.; Hussain, I.; Momotaj, H.; van Geen, A.; Howe, G.R.; Ahsan, H. Modification of risk of arsenic-induced skin lesions by sunlight exposure, smoking, and occupational exposures in Bangladesh. Epidemiology 2006, 17, 459–467. [Google Scholar] [CrossRef]
- Argos, M.; Kalra, T.; Pierce, B.L.; Chen, Y.; Parvez, F.; Islam, T.; Ahmed, A.; Hasan, R.; Hasan, K.; Sarwar, G.; et al. A prospective study of arsenic exposure from drinking water and incidence of skin lesions in Bangladesh. Am. J. Epidemiol. 2011, 174, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011, 2011, 431287. [Google Scholar] [CrossRef]
- Smith, A.H.; Lingas, E.O.; Rahman, M. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bull. World Health Organ. 2000, 78, 1093–1103. [Google Scholar]
- Kobayashi, Y.; Agusa, T. Arsenic Metabolism and Toxicity in Humans and Animals: Racial and Species Differences. In Arsenic Contamination in Asia: Biological Effects and Preventive Measures; Yamauchi, H., Sun, G., Eds.; Springer: Singapore, 2019; pp. 13–28. [Google Scholar] [CrossRef]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, W.R. Chemical Mechanism of Arsenic Biomethylation. Chem. Res. Toxicol. 2014, 27, 457–461. [Google Scholar] [CrossRef]
- Buchet, J.P.; Lauwerys, R.; Roels, H. Comparison of the urinary excretion of arsenic metabolites after a single oral dose of sodium arsenite, monomethylarsonate, or dimethylarsinate in man. Int. Arch. Occup Environ. Health 1981, 48, 71–79. [Google Scholar] [CrossRef]
- Martin, E.M.; Styblo, M.; Fry, R.C. Genetic and epigenetic mechanisms underlying arsenic-associated diabetes mellitus: A perspective of the current evidence. Epigenomics 2017, 9, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Argos, M. Arsenic Exposure and Epigenetic Alterations: Recent Findings Based on the Illumina 450K DNA Methylation Array. Curr. Environ. Health Rep. 2015, 2, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Reichard, J.F.; Puga, A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2010, 2, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Eyvani, H.; Moghaddaskho, F.; Kabuli, M.; Zekri, A.; Momeny, M.; Tavakkoly-Bazzaz, J.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Arsenic trioxide induces cell cycle arrest and alters DNA methylation patterns of cell cycle regulatory genes in colorectal cancer cells. Life Sci. 2016, 167, 67–77. [Google Scholar] [CrossRef]
- Ajees, A.A.; Rosen, B.P. As(III) S-adenosylmethionine methyltransferases and other arsenic binding proteins. Geomicrobiol. J. 2015, 32, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Hamdi, M.; Yoshinaga, M.; Packianathan, C.; Qin, J.; Hallauer, J.; McDermott, J.R.; Yang, H.C.; Tsai, K.J.; Liu, Z. Identification of an S-Adenosylmethionine (SAM) dependent arsenic methyltransferase in Danio rerio. Toxicol. Appl. Pharmacol. 2012, 262, 185–193. [Google Scholar] [CrossRef]
- Singh, A.P.; Goel, R.K.; Kaur, T. Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 2011, 18, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caumette, G.; Koch, I.; Reimer, K.J. Arsenobetaine formation in plankton: A review of studies at the base of the aquatic food chain. J. Environ. Monit. 2012, 14, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Hubaux, R.; Becker-Santos, D.D.; Enfield, K.S.; Rowbotham, D.; Lam, S.; Lam, W.L.; Martinez, V.D. Molecular features in arsenic-induced lung tumors. Mol. Cancer 2013, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, V.N.; Verschuuren, M.; van Oers, K.; Espin, S.; Sanchez-Virosta, P.; Eeva, T.; Ruuskanen, S. Does Arsenic Contamination Affect DNA Methylation Patterns in a Wild Bird Population? An Experimental Approach. Environ. Sci. Technol. 2021, 55, 8947–8954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yang, Y.; Wang, X.; Chong, Z.; Yin, R.; Song, S.H.; Zhao, C.; Li, C.; Huang, H.; Sun, B.F.; et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucleic Acids Res. 2014, 42, 1593–1605. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, J.; Li, L.; Amato, N.J.; Wang, Z.; Wang, Y. Arsenite Targets the Zinc Finger Domains of Tet Proteins and Inhibits Tet-Mediated Oxidation of 5-Methylcytosine. Environ. Sci. Technol. 2015, 49, 11923–11931. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N(6)-methyladenine: A new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705–710. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Wion, D.; Casadesus, J. N6-methyl-adenine: An epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192. [Google Scholar] [CrossRef]
- O’Brown, Z.K.; Greer, E.L. N6-Methyladenine: A Conserved and Dynamic DNA Mark. Adv. Exp. Med. Biol. 2016, 945, 213–246. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin 2015, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef]
- Ehrlich, M.; Wang, R.Y. 5-Methylcytosine in eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Dore, L.C.; et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef] [Green Version]
- Beh, L.Y.; Debelouchina, G.T.; Clay, D.M.; Thompson, R.E.; Lindblad, K.A.; Hutton, E.R.; Bracht, J.R.; Sebra, R.P.; Muir, T.W.; Landweber, L.F. Identification of a DNA N6-Adenine Methyltransferase Complex and Its Impact on Chromatin Organization. Cell 2019, 177, 1781–1796. [Google Scholar] [CrossRef]
- Luo, G.Z.; Hao, Z.; Luo, L.; Shen, M.; Sparvoli, D.; Zheng, Y.; Zhang, Z.; Weng, X.; Chen, K.; Cui, Q.; et al. N(6)-methyldeoxyadenosine directs nucleosome positioning in Tetrahymena DNA. Genome Biol. 2018, 19, 200. [Google Scholar] [CrossRef]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizabal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Huang, H.; Liu, D.; Cheng, Y.; Liu, X.; Zhang, W.; Yin, R.; Zhang, D.; Zhang, P.; Liu, J.; et al. N6-methyladenine DNA modification in Drosophila. Cell 2015, 161, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Koziol, M.J.; Bradshaw, C.R.; Allen, G.E.; Costa, A.S.H.; Frezza, C.; Gurdon, J.B. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct Mol. Biol. 2016, 23, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhou, C.; Yuan, Q. Role of DNA and RNA N6-Adenine Methylation in Regulating Stem Cell Fate. Curr. Stem Cell Res. Ther. 2018, 13, 31–38. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, T.P.; Gimple, R.C.; Li, Z.; Prager, B.C.; Wu, Q.; Yu, Y.; Wang, P.; Wang, Y.; Gorkin, D.U.; et al. N(6)-methyladenine DNA Modification in Glioblastoma. Cell 2018, 175, 1228–1243 e1220. [Google Scholar] [CrossRef] [Green Version]
- Yao, B.; Cheng, Y.; Wang, Z.; Li, Y.; Chen, L.; Huang, L.; Zhang, W.; Chen, D.; Wu, H.; Tang, B.; et al. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun. 2017, 8, 1122. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Niu, R.; Huang, T.; Shao, L.W.; Peng, Y.; Ding, W.; Wang, Y.; Jia, G.; He, C.; Li, C.Y.; et al. N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell Biol. 2019, 21, 319–327. [Google Scholar] [CrossRef]
- Iyer, L.M.; Zhang, D.; Aravind, L. Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays 2016, 38, 27–40. [Google Scholar] [CrossRef]
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N(6)-Methyladenine DNA Modification in the Human Genome. Mol. Cell 2018, 71, 306–318 e307. [Google Scholar] [CrossRef]
- Kweon, S.M.; Chen, Y.; Moon, E.; Kvederaviciute, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N(6)-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 2019, 74, 1138–1147 e1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.H.; Yang, S.; Wei, J.; Shea, C.R.; Zhong, W.; Wang, F.; Shah, P.; Kibriya, M.G.; Cui, X.; Ahsan, H.; et al. Autophagy of the m(6)A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat. Commun. 2021, 12, 2183. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, S.; Cui, Y.H.; Wei, J.; Shah, P.; Park, G.; Cui, X.; He, C.; He, Y.Y. METTL14 facilitates global genome repair and suppresses skin tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2025948118. [Google Scholar] [CrossRef]
- Weinmuellner, R.; Kryeziu, K.; Zbiral, B.; Tav, K.; Schoenhacker-Alte, B.; Groza, D.; Wimmer, L.; Schosserer, M.; Nagelreiter, F.; Rosinger, S.; et al. Long-term exposure of immortalized keratinocytes to arsenic induces EMT, impairs differentiation in organotypic skin models and mimics aspects of human skin derangements. Arch. Toxicol. 2018, 92, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Liu, D.; Wang, Z.; Tian, R.; Zuo, Y. Multi-substrate selectivity based on key loops and non-homologous domains: New insight into ALKBH family. Cell Mol. Life Sci 2021, 78, 129–141. [Google Scholar] [CrossRef]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem 2015, 290, 20734–20742. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falnes, P.O.; Bjoras, M.; Aas, P.A.; Sundheim, O.; Seeberg, E. Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res. 2004, 32, 3456–3461. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, M.; Eckstein, M.; Eleazer, R.; Smith, C.; Fondufe-Mittendorf, Y.N. Genome-wide DNA methylation reprogramming in response to inorganic arsenic links inhibition of CTCF binding, DNMT expression and cellular transformation. Sci. Rep. 2017, 7, 41474. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K.; Zhitkovich, A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol. 2008, 21, 28–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, D.; Shinkai, Y.; Kumagai, Y. Signal transduction pathways and transcription factors triggered by arsenic trioxide in leukemia cells. Toxicol. Appl. Pharmacol. 2010, 244, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Reichard, J.F.; Schnekenburger, M.; Puga, A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 2007, 352, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Y.; Xu, W.; Dong, L.; Guo, Y.; Bi, K.; Zhu, C. Arsenic trioxide inhibits DNA methyltransferase and restores TMS1 gene expression in K562 cells. Acta Haematol. 2015, 133, 18–25. [Google Scholar] [CrossRef]
- Shen, C.; Yan, T.; Tong, T.; Shi, D.; Ren, L.; Zhang, Y.; Zhang, X.; Cao, Y.; Yan, Y.; Ma, Y.; et al. ALKBH4 Functions as a Suppressor of Colorectal Cancer Metastasis via Competitively Binding to WDR5. Front. Cell Dev. Biol. 2020, 8, 293. [Google Scholar] [CrossRef]
- Jingushi, K.; Aoki, M.; Ueda, K.; Kogaki, T.; Tanimoto, M.; Monoe, Y.; Ando, M.; Matsumoto, T.; Minami, K.; Ueda, Y.; et al. ALKBH4 promotes tumourigenesis with a poor prognosis in non-small-cell lung cancer. Sci. Rep. 2021, 11, 8677. [Google Scholar] [CrossRef]
- Li, M.M.; Nilsen, A.; Shi, Y.; Fusser, M.; Ding, Y.H.; Fu, Y.; Liu, B.; Niu, Y.; Wu, Y.S.; Huang, C.M.; et al. ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nat. Commun. 2013, 4, 1832. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.-H.; Wilkinson, E.; Peterson, J.; He, Y.-Y. ALKBH4 Stabilization Is Required for Arsenic-Induced 6mA DNA Methylation Inhibition, Keratinocyte Malignant Transformation, and Tumorigenicity. Water 2022, 14, 3595. https://doi.org/10.3390/w14223595
Cui Y-H, Wilkinson E, Peterson J, He Y-Y. ALKBH4 Stabilization Is Required for Arsenic-Induced 6mA DNA Methylation Inhibition, Keratinocyte Malignant Transformation, and Tumorigenicity. Water. 2022; 14(22):3595. https://doi.org/10.3390/w14223595
Chicago/Turabian StyleCui, Yan-Hong, Emma Wilkinson, Jack Peterson, and Yu-Ying He. 2022. "ALKBH4 Stabilization Is Required for Arsenic-Induced 6mA DNA Methylation Inhibition, Keratinocyte Malignant Transformation, and Tumorigenicity" Water 14, no. 22: 3595. https://doi.org/10.3390/w14223595