
Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components
by
 , ,
, ,
Jean-François Rontani
1,*,
Bruno Charriere
2,3,
Christophe Menniti
2,3,
Itzhak Katra
4 and
Dominique Aubert
2,3 1
Mediterranean Institute of Oceanography (MIO), Aix Marseille University, Université de Toulon, CNRS, IRD, MIO UM 110, 13288 Marseille, France
2
Centre de Formation et de Recherche sur les Environnements Méditerranéens, UMR 5110, Centre National de la Recherche Scientifique (CNRS), 52 Avenue Paul Alduy, 66860 Perpignan, France
3
Centre de Formation et de Recherche sur les Environnements Méditerranéens, UMR 5110, Université de Perpignan Via Domitia, 52 Avenue Paul Alduy, 66860 Perpignan, France
4
Department of Geography and Environmental Development, Ben-Gurion University of the Negev, P.O. Box 653, Beersheba 84105, Israel
*
Author to whom correspondence should be addressed.
Water 2022, 14(24), 4092; https://doi.org/10.3390/w14244092
Submission received: 27 September 2022
/
Revised: 6 December 2022
/
Accepted: 13 December 2022
/
Published: 15 December 2022
(This article belongs to the Special Issue Impact of Atmospheric and River Inputs on the Transfer of Elements and Organic Matter to the Ocean)
Abstract
:Lipids and their oxidation products were quantified in loess samples from the Negev Desert (Israel), well known to be a source of desert dusts in the eastern Mediterranean Basin. The results obtained showed the presence of higher plant material (angiosperms and gymnosperms), but also bacteria and fungi. Although a strong autoxidation of lipids could be demonstrated, the resulting oxidation products appeared to be weakly accumulated, likely due to the high temperatures and solar irradiance observed in the Negev Desert. Incubation of this dust analogue in fresh water (to mimic their behavior in rainwater) resulted in the release of metal ions (mainly iron), but also a fast heterolytic degradation of their weak content of hydroperoxides. Induction of autoxidation processes in dry and wet atmospheric dusts arising from the Negev Desert in seawater (needing simultaneous presence of metal ions and hydroperoxides) seems thus very unlikely due to the relatively high pH of seawater hindering metal dissolution and the degradation of hydroperoxides in rainwater.
1. Introduction
Terrestrial particulate organic matter (TPOM) discharged by rivers and atmospheric inputs into the sea partly consists of previously-degraded nitrogen-poor terrestrial plant residues and has long been considered refractory to marine decomposition processes [1,2]. However, coastal marine sediments show a weak terrestrial signature [3], suggesting that either the global budgets and distribution estimates are wrong or, more likely, that TPOM rapidly undergoes intensive degradation once at sea [4]. The processes involved in the degradation of these very large quantities of TPOM at sea remain poorly understood, which is a bottleneck to the development of coupled physical–biogeochemical numerical models that could correctly estimate the fate of TPOM in the oceans and its implications for trophic balance of marine ecosystems.
The paradigm of TPOM being refractory to decomposition processes at sea has recently been challenged by several studies conducted in Siberian [4,5] and Mediterranean estuaries [6]. These studies have demonstrated that, contrary to what was thought, TPOM is in fact very sensitive to bacterial degradation processes at sea. More recently, research in the Mackenzie, Rhône and Amazon Deltas used specific lipid markers [6] to demonstrate that well-preserved TPOM in rivers underwent very intense degradation as soon as it reached the sea [6,7,8,9,10]. These works have shown that the degradation was driven by two major processes: bacterial degradation and autoxidation.
Autoxidation processes have been totally ignored in the marine environment until now. Autoxidation is an abiotic oxidation of OM involving radical chain reactions in three phases: (i) initiation of free radicals, (ii) propagation as these radicals attack other molecules, and (iii) termination once the reactive species disappear. These processes, which act in any oxic environment, can affect not only unsaturated lipids (see [11] for a review) but also proteins or amino acids (see [12] for a review) and biopolymers such as lignin [13]. We previously showed that the strong autoxidation of TPOM observed in estuaries resulted from the intervention of certain radical-producing enzymes (lipoxygenases) contained in plant debris and whose activity intensifies with increasing salinity [9], and that the intensity of this abiotic degradation in high-, mid- and low-latitude areas was related to the photooxidation or autoxidation status (and thus hydroperoxide contents) of higher plants on Earth [9].
We also attributed the high biodegradation of TPOM observed in estuaries to: (i) the selection of bacteria better adapted to degrading TPOM in the salinity gradient [10] and (ii) the intervention of a “priming effect” that favors assimilation of recalcitrant substrates (TPOM) in the presence of readily-assimilable cosubstrates (phytoplankton) [14].
In the more general framework of our studies of the degradation of TPOM at sea, we focused on the aeolian inputs from deserts. Deserts are effectively the biggest source of dust atmospheric inputs, with an estimate of 600 Tg yr−1 from the Sahara alone [15,16]. These aeolian particles, composed mainly of mineral material, contain a number of metals (including Cu, Fe, Mn, Zn, Co, and Cd) but also fragments of leaves or cuticles of C4 plants (halophytes and various grasses) that dominate the sparse sub-Saharan vegetation [17]. These desert inputs reach the sea surface either as dust (dry inputs) or as rain (wet inputs). It was recently shown that the dissolution of metal components (especially iron) carried by dry inputs is extremely low in seawater but relatively high in rainwater due to its relative acidity [18]. Once dissolved, the metal components of these inputs are likely to catalyze (via redox reactions [19]) the autoxidation of the TPOM they carry. Once inputs come into contact with seawater, these autoxidative processes could also be induced by lipoxygenases found in higher plant debris and whose activity intensifies in the presence of NaCl [9].
Here, to investigate these processes, we quantified lipids and their oxidation products in dust analogue by loess soil from the Negev Desert (Israel), which is a well-known source of desert dust in the Eastern Mediterranean Basin [20]. We also performed lipid and metal analyses after first incubating these samples in slightly acidic fresh water (to mimic their behavior in rain). These different experiments converge to afford a better understanding of the fate of dry and wet atmospheric inputs in the Mediterranean Sea.
2. Materials and Methods
2.1. Sampling and Chemical Characterization of the Dust Analogue Material
The desert dust analogue was produced from loess soil in the southern Negev Desert in Israel. This region is covered by widespread Pleistocene loess material that has since been shifted by fluvial and aeolian processes. The sampling site (31°14′48.43″ N, 34°38′2.97″ E) is situated within a closed area (a military base) without any human interference during the last decades. This soil is characterized by bare surfaces as well as coverage of biological crust, and annual and seasonal vegetation. More information and photos about these plots and about dust emission can be found in [21] and [22].
Samples were extracted from the topsoil layer (2 cm) of bare surfaces. This layer is subjected to dust emission, as demonstrated by [21]. Total organic carbon and carbonate content are 2.3% and 22.4%, respectively [21]. To produce the dust analogue, we followed common procedures described by previous works on dust analogue from the loess soil (e.g., [23,24,25,26]). The samples were processed through a setup of sieves to achieve a particle size smaller than 63 µm, which is the threshold for aeolian transport of sediments (e.g., [27]). Mineralogical composition of the dust analogue by the XRD method revealed 42% quartz, 27% calcite, 20% Na silicate, 3% K silicate, 6% Kaolinite, and 2% Fe2O3. Particle size distribution by the laser diffractor method revealed that the fraction of the clay-sized particle (<2 µm) is 13.69%, and fine silt (2–20 µm) is 39.59%. These properties are comparable to those of Middle Eastern dust samples that were previously captured in Israel (e.g [22,28,29,30,31,32]) as discussed also in [26].
2.2. Major and Trace Metal Composition of the Loessic Soil
Trace metals were analyzed in duplicate after total acid digestion. Briefly, homogenized samples were transferred into acid-cleaned Teflon tubes added with 1.5 mL of a HNO3/H2O2 (1:2) mixture to remove organic matter. After drying, the mineral fraction was digested by adding 1 mL of HF/HNO3 (1:1 trace-metal grade) at 100 °C for 12 h. After re-drying, the samples were held diluted in a HNO3 2% solution until analysis [33]. Major and trace elements were measured by inductively-coupled plasma mass spectrometry on an Agilent 7700x system.
2.3. Incubations in Fresh Water
Crustal dust dissolution in rainwater is hard to simulate, as it depends on a cluster of factors such as rain pH [34,35,36]; aging of the dust over long-range transport [37], including in cloud acidification processes [38]; dust concentration in the atmosphere and composition of the dust itself [39]. Consequently, the main objective we set here was not to faithfully mimic those processes leading to metal release by dust dissolution, but rather to get a rough idea of the minimal metal concentration that can be expected through rainwater dissolution. We therefore decided to run dissolution experiments without any pre-treatment (contrary to previous tests [40]) in slightly acidic mQ water at a pH around 6.8 (i.e., close to values recorded for rainwater in Israel [41]).
One hundred milligrams of dust analogue was incubated in 100 mL of ultra-pure mQ water at pH 6.82 at 20 °C in the dark in duplicate. This concentration is in the range of those measured in wet atmospheric inputs (5–8000 mg L−1) in the Mediterranean Sea [42]. Four incubation times were selected: T0 (dust particles removed immediately after shaking), T12h, T24h and T96h. After incubation, the solutions were decanted and the supernatant was filtered on nitrocellulose (Whatman 47 mm, 0.45-µm-pore-size membrane filter). The dissolved fraction was acidified to 2% final concentration using nitric acid (TraceSELECT, Fluka). The particles were recovered on pre-combusted GF/F filters and stored at −20 °C until lipid analysis. All the material used was pre-washed in HCl 1 M solution (Trace SELECT, Fluka) and then rinsed three times with ultra-pure mQ water.
2.4. Treatment
Samples (dust analogue or GF/F filters) were reduced at room temperature with excess NaBH4 (70 mg) after adding methanol (25 mL, 30 min) to reduce labile hydroperoxides (resulting from photo- or autoxidation) to alcohols, which are more amenable to analysis by gas chromatography (GC). Water (25 mL) and KOH (2.8 g) were then added, and the resulting mixture was saponified by refluxing (2 h). After cooling, the mixture was acidified (HCl, 2 N) to pH 1 and extracted with dichloromethane (DCM; 3 × 20 mL). The combined DCM extracts were dried over anhydrous Na2SO4, filtered, and concentrated by rotary evaporation at 40 °C to yield total lipid extracts (TLEs).
A different treatment was employed to estimate the relative proportions of hydroperoxides and their ketonic and alcoholic degradation products in atmospheric inputs [43]. The procedure involved ultrasonic extraction of lipids with chloroform–methanol–water (1:2:0.8, v/v/v), separation of the supernatant by centrifugation at 3500 g, evaporation to dryness, and division of the residue into two equal parts. The first sub-sample was acetylated in acetic anhydride–pyridine (1:2, v/v) overnight, which converted hydroperoxides to the corresponding ketones [44], then evaporated to dryness and saponified. The second sub-sample was evaporated to dryness, reduced with NaBD4, and saponified. The amounts of hydroxyacids present after acetylation and NaBD4 reduction were then compared in order to estimate the amounts of hydroperoxyacids and hydroxyacids present in the samples, and deuterium labelling (via NaBD4 reduction) made it possible to estimate the proportion of ketoacids.
2.5. Silylation
Dry TLEs and standards were derivatized by dissolving them in 300 µL pyridine/bis-(trimethylsilyl)trifluoroacetamide (BSTFA; Supelco; 2:1, v/v) and silylating them in a heating block (50 °C, 1 h). After evaporation to dryness under a stream of N2, the derivatized residue was dissolved in ethyl acetate/BSTFA (to avoid desilylation) and then analyzed by GC-MS/MS and GC-QTOF.
2.6. Gas Chromatography–Electron Ionization Quadrupole Time-of-Flight Mass Spectrometry (GC-QTOF)
Accurate mass measurements were made in full scan mode with use of an Agilent 7890B/7200 GC/QTOF system (Agilent Technologies, Les Ulis, France) with a cross-linked 5% phenyl-methylpolysiloxane capillary column (Agilent Technologies; HP-5MS Ultra inert, 30 m × 0.25 mm, 0.25-µm film thickness). Analyses were performed with an injector set at 270 °C and operating in pulsed splitless mode. Oven temperature was ramped from 70 °C to 130 °C at 20 °C min−1 and then to 300 °C at 5 °C min−1. Pressure of the Carrier gas (He) was held at 0.69 × 105 Pa until the end of the temperature program. Instrument temperatures were 300 °C for the transfer line and 230 °C for the ion source. Nitrogen (1.5 mL min−1) was used as collision gas. Accurate mass spectra were recorded across the range m/z 50–700 at 4 GHz with the collision gas opened. The QTOF-MS instrument provided a typical resolution ranging from 8009 to 12,252 from m/z 68.9955 to 501.9706. Perfluorotributylamine (PFTBA) was used for daily MS calibration. Unsaturated lipid components of higher plants (erucic, linoleic, p-coumaric, and ferulic acids; sitosterol; squalene; and α- and β-amyrins) were identified by comparing their TOF mass spectra, accurate masses and retention times against standards. Quantification of each compound involved extraction of specific accurate fragment ions, peak integration, and determination of individual response factors using external standards and Mass Hunter software (Agilent Technologies, Les Ulis, France).
2.7. Gas Chromatography/Tandem Mass Spectrometry
GC–MS/MS analyses were performed using an Agilent 7890A/7010 tandem quadrupole gas chromatograph system (Agilent Technologies, Les Ulis, France) with a cross-linked 5% phenyl-methylpolysiloxane capillary column (Agilent; HP-5MS ultra inert, 30 m × 0.25 mm, 0.25-µm film thickness). Analyses were performed with an injector set at 270 °C operating in pulsed splitless mode. Oven temperature was ramped from 70 °C to 130 °C at 20 °C min−1, then to 250 °C at 5 °C min−1 and finally to 300 °C at 3 °C min−1. Pressure of the carrier gas (He) was held at 0.69 × 105 Pa until the end of the temperature program and then ramped from 0.69 × 105 Pa to 1.49 × 105 Pa at 0.04 × 105 Pa min−1. The mass spectrometry conditions were: electron energy, 70 eV; source temperature, 230 °C; quadrupole 1 temperature, 150 °C; quadrupole 2 temperature, 150 °C; collision gas (N2) flow rate, 1.5 mL min−1; quench gas (He) flow rate, 2.25 mL min−1; mass range, 50–700 daltons; cycle time, 313 ms. Oxidation products of monounsaturated fatty acids (MUFAs), sterols and amyrins were assigned by comparing retention times and mass spectra against standards. Quantification was carried out with external standards in multiple reaction monitoring (MRM) mode. Precursor ions were selected from the more intense ions (and specific fragmentations) observed in electron ionization (EI) mass spectra [45].
2.8. Metal Analyses
Major and trace element concentrations were measured by inductively-coupled plasma mass spectrometry (ICP-MS) on an Agilent 7000x system. External calibration was performed using commercial solutions (Carlo Erba Company, Val de Reuil, France) diluted as required in 2% nitric acid. Blanks were lower than 2% for all elements.
3. Results and Discussion
3.1. Composition of Dust Analogue
3.1.1. Organic Composition
Negev loess deposits originate from the late Pleistocene [46]. Due to the lack of dating, it was not possible to differentiate the organic compounds originating from the vegetation of that period from those of today. TLEs of this dust analogue appeared to be dominated by unspecific C14-C18 saturated fatty acids and C18:1Δ9 (oleic) acid (Table 1 and Table S1). They also contained small proportions of more specific acids such as C18:1Δ11 (vaccenic) and branched C15:0 fatty acids indicative of the presence of bacteria [47,48]. The presence of ω, (9-10)-dihydroxy-C16:0 acids (components of plant cuticular waxes [49,50]), ferulic and p-coumaric acids (components of lignocellulose, [51]), sitosterol and campesterol (dominant sterols of higher plants, [52]) (Table 1) attests to the presence of higher plant material in these loess deposits.
More precise information concerning the nature of this plant material was then obtained using more specific markers such as C22:1Δ13 (erucic) acid, dehydroabietic acid and α- and β-amyrins (Table 1), indicating the presence of Brassicasseae [53], gymnosperms [54,55] and angiosperms [56], respectively. Methoxyhydrins, diols and chlorohydrins resulting from the degradation of 9,10-epoxyoctadecanoic acid during the treatment [49] were also detected in the TLEs (Table 1). The presence of this epoxy acid, formed from oleic acid by fungal enzymes in wheat plants infected by stem rust [57], confirmed the presence of fungi in the loess samples investigated.
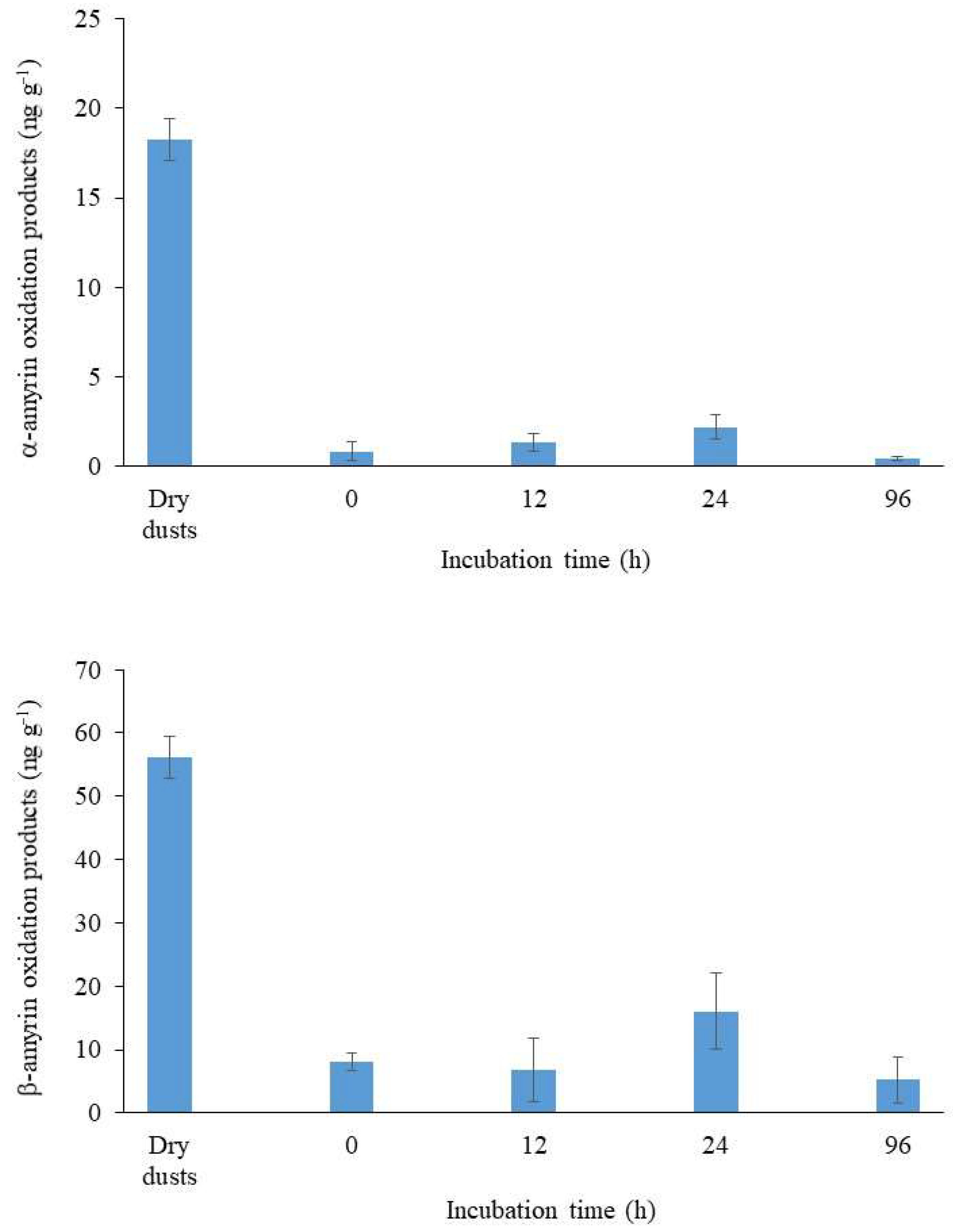
Photo- and autoxidation products of MUFAs (isomeric allylic hydroxy acids [58]), sterols (3β,6α/β-dihydroxysterols and 3β,5α,6β-steratriols [7]), dehydroabietic acid (7α/β−hydroxydehydroabietic acids [59]) and amyrins (11-oxoamyrins [60]) were quantified after NaBH4-reduction. The results obtained (Figure 1, Table S2) showed very low rates of oxidation of MUFAs, sterols and dehydroabietic acid.
The surprising preservation of higher plant tracers (erucic acid, campesterol, sitosterol and dehydroabietic acid) can be attributed to: (i) the presence of high proportions of ferulic and p-coumaric acids in this material (Table 1) exhibiting highly efficient antioxidant activity due to their ability to scavenge free radicals, donate hydrogen atoms or electrons, or chelate metal cations [61,62] or (ii) an intense degradation of the oxidation products formed (hydroperoxides) under the prevailing environmental conditions (strong UV irradiance and high temperatures of the Negev desert). The strong oxidation observed in the case of α- and β-amyrins (Figure 1, Table S2) argues strongly for the second hypothesis: autoxidation of amyrins affords particularly stable allylic 11α-hydroperoxyamyrins that resist NaBH4 reduction and are only converted to 11-oxoamyrins at the very high temperature (270 °C) of splitless GC injectors [60]. The high proportions of these resilient oxidation products detected in the samples (Figure 1) thus attest to the presence of strongly altered angiosperm material. The low proportions of oxidation products of the other unsaturated higher plant lipids (Figure 1) thus likely result from the lability of the hydroperoxides formed rather than from any lack of reactivity of these lipids. Note that the higher oxidation state of α-amyrin relative to β-amyrin is not in agreement with previous observations carried out in senescent higher plant material [60].
3.1.2. Metal Concentrations
Concentrations of trace metals of concern in this study (Fe, V, Mn, Zn, Cu, Co and Cd), as well as major elements allowing to characterize the original loess material (Ca and Al), are presented in Table 2 (dust sample). Two replicates were analyzed. Total Ca and Al concentrations were about 13.8% and 4.1% in the sample, respectively. The previous determination of carbonate content at this site (around 22%) by [63] would indicate that part of the Ca content could originate from a source different from calcareous rock (detrital siliciclastic rocks or an organic source). Ref. [64] evidenced fairly high compositional variability in Negev loess. Calcite content ranged from 16% to 45%. Quartz was fairly dominant, but the bulk loess composition also featured phyllosilicates, K-feldspars and plagioclase. Furthermore, the coarse fraction of the loess is essentially quartzofeldspathic, which explains the fairly high Al concentration, whereas Fe concentration was lower than that typically measured in the upper continental crust [65].
There are scarce data in the literature regarding trace metals present in loess samples from Israel. Nevertheless, one can mention Netivot loess [28], located a few kilometers north, which shows a quite similar composition. Only Zn and Cd contents in our loess sample are slightly more concentrated than in the Netivot one (65 µg.g−1 and 0.63 µg.g−1, respectively). Moreover, Lucke et al. [66] reported that concentrations of Zn and V in loessial soil, Negev reference samples, and recent dust storms occurring in the Negev area ranged between 43 and 98 µg.g−1 and 14 and 115 µg.g−1, respectively. These reported values fit well with the contents observed in our sample.
3.2. Incubations of Dust Analogue in Fresh Water
3.2.1. Metal Dissolution
Fe, Mn, Co and, to a lesser extent, V exhibited quite similar patterns during incubation, with an increase in concentration at T12h and T24h followed by a decrease at the final time point T96h (Figure 2). Their concentrations peaked at T24h to reach 26-fold (Fe), 6-fold (Mn), 13-fold (Co) and 6-fold (V) higher that at the initial time point T0. Interestingly, Fe and Mn concentrations showed huge decreases at the final T96h time point (63% and 70%, respectively) compared to the initial T0 concentration, whereas Cu, Cd and Zn immediately showed a high solubilization rate that stabilized with time. Among these three elements, only Zn showed a significant decrease in concentration at T96h. Note that the standard deviation for this Zn was relatively high, indicating heterogeneous dissolution processes from one sample to another. Nevertheless, the highest release percentage relative to the metal concentrations in dusts (Table 2) was for Zn (about 15% at T0), followed by Cu (4.4% at T96h), then Cd, V, Co, Al, Fe and Mn (2.89%, 2.73%, 1.91%, 1.84%, 1.55% and 1.29% at T96h, respectively).
It has been shown previously that metals fall into three modes of dissolution: gradually-dissolving metals that exhibit increasing dissolved concentrations over time (Ni, Al, Cu and, to a lesser extent, Mn); rapidly-dissolving metals for which all soluble metal gets released immediately and remains quite constant over time (Zn, Co, Cd); and particle-reactive metals for which concentrations first increase but then decrease due to sorption onto particles or container walls (Fe) [67]. This fits partially with our observations. High Zn dissolution could also originate from recent soluble atmospheric anthropogenic Zn deposited onto the soil sample [66]. These incubation experiments highlight the possible release of metals potentially involved in autoxidation processes during atmospheric transport.
3.2.2. Impact on Lipid Oxidation Products
If, as we confirmed above, the incubation of dust analogue in fresh water leads to the dissolution of certain metal ions, it could also affect the stability of the hydroperoxides present in the dust material, which are likely to play a key role in inducing autoxidative processes at sea [45]. Indeed, the relative acidity of rain water (up to pH 2 in highly-polluted zones [18]) can also induce quick hydrolytic cleavage of these compounds [68].
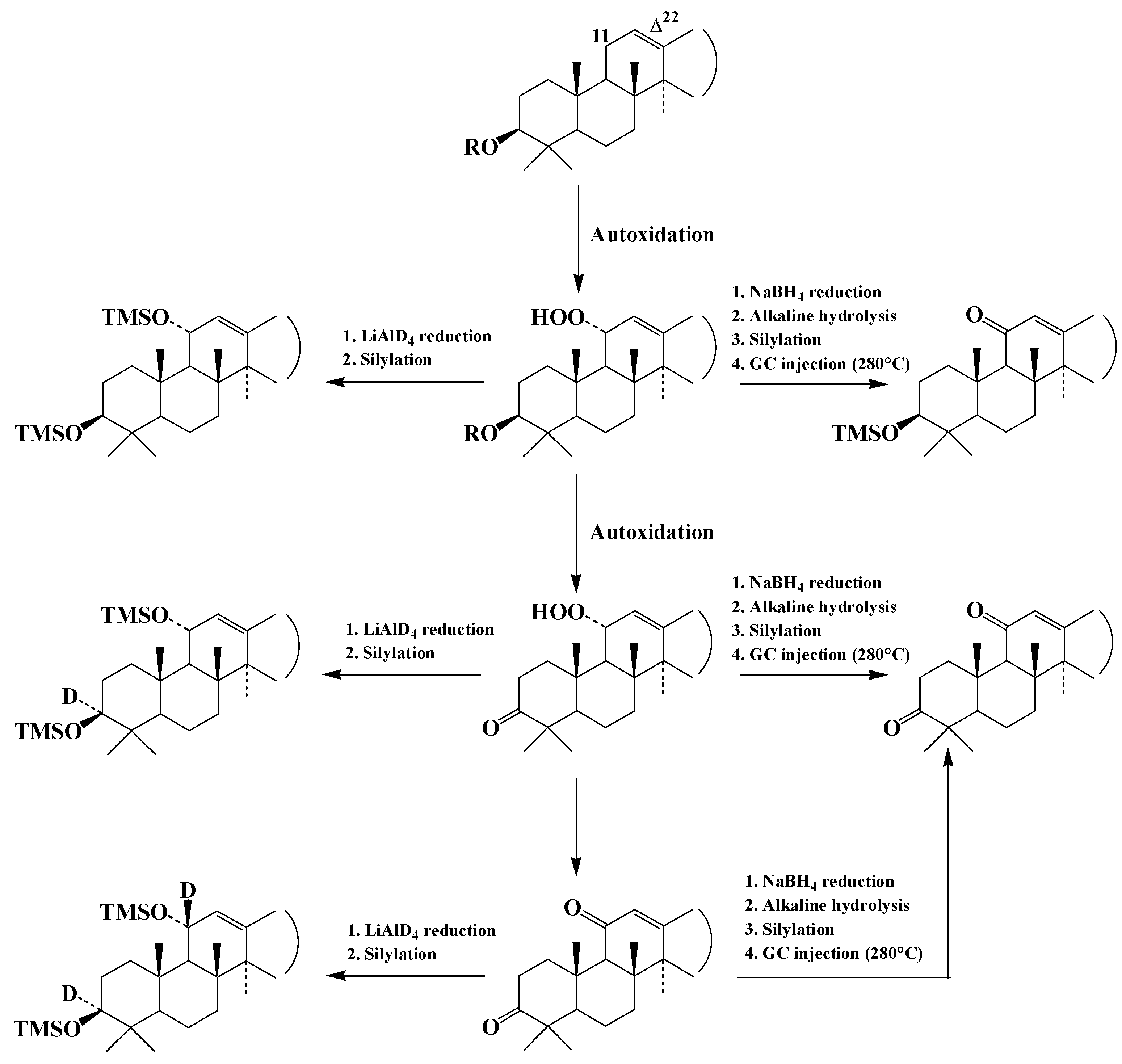
We thus ran analyses to track degradation of the more resistant hydroperoxides (i.e., oxidation products of amyrins) in fresh water. In addition to 11α-hydroperoxyamyrins, intensive autoxidation of α- and β-amyrins also produces 11α-hydroperoxyamyrons and 11-oxoamyrons [48] (Figure 3). NaBH4 reduction, which is inefficient on the hydroperoxy and oxo groups of these different compounds, was unable to differentiate 11α-hydroperoxyamyrins from 11-oxoamyrins or 11α-hydroperoxyamyrons from 11-oxoamyrons (Figure 3). In order to provide proof that the focal compounds were 11α-hydroperoxyamyrins and 11α-hydroperoxyamyrons, TLEs were reduced with LiAlD4 and silylated (Figure 3).
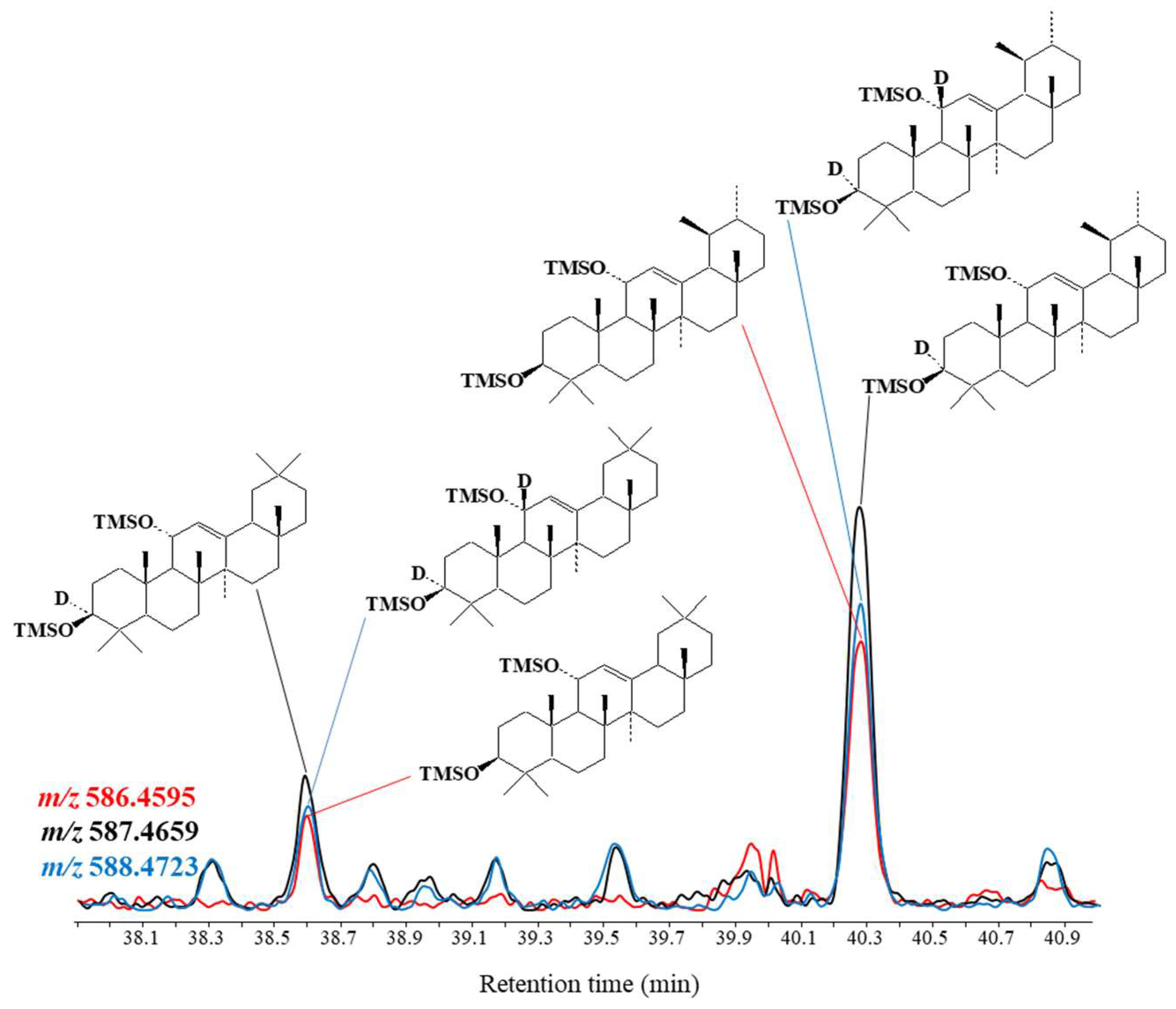
The results obtained (Figure 4) showed that 70% of the amyrin oxidation products in Negev loess deposits are in the form of 11α-hydroperoxyamyrins and 11α-hydroperoxyamyrons, with the remaining 30% in the form of 11-oxoamyrons.
Incubating Negev loess deposits in fresh water resulted in very fast degradation of amyrin oxidation products (Figure 5, Table S3), likely from heterolytic cleavage of 11α-hydroperoxyamyrins and 11α-hydroperoxyamyrons. While this heterolysis of the hydroperoxide O–O bond is generally acid-catalyzed [68,69], it was previously observed in senescent phytoplanktonic cells [70].
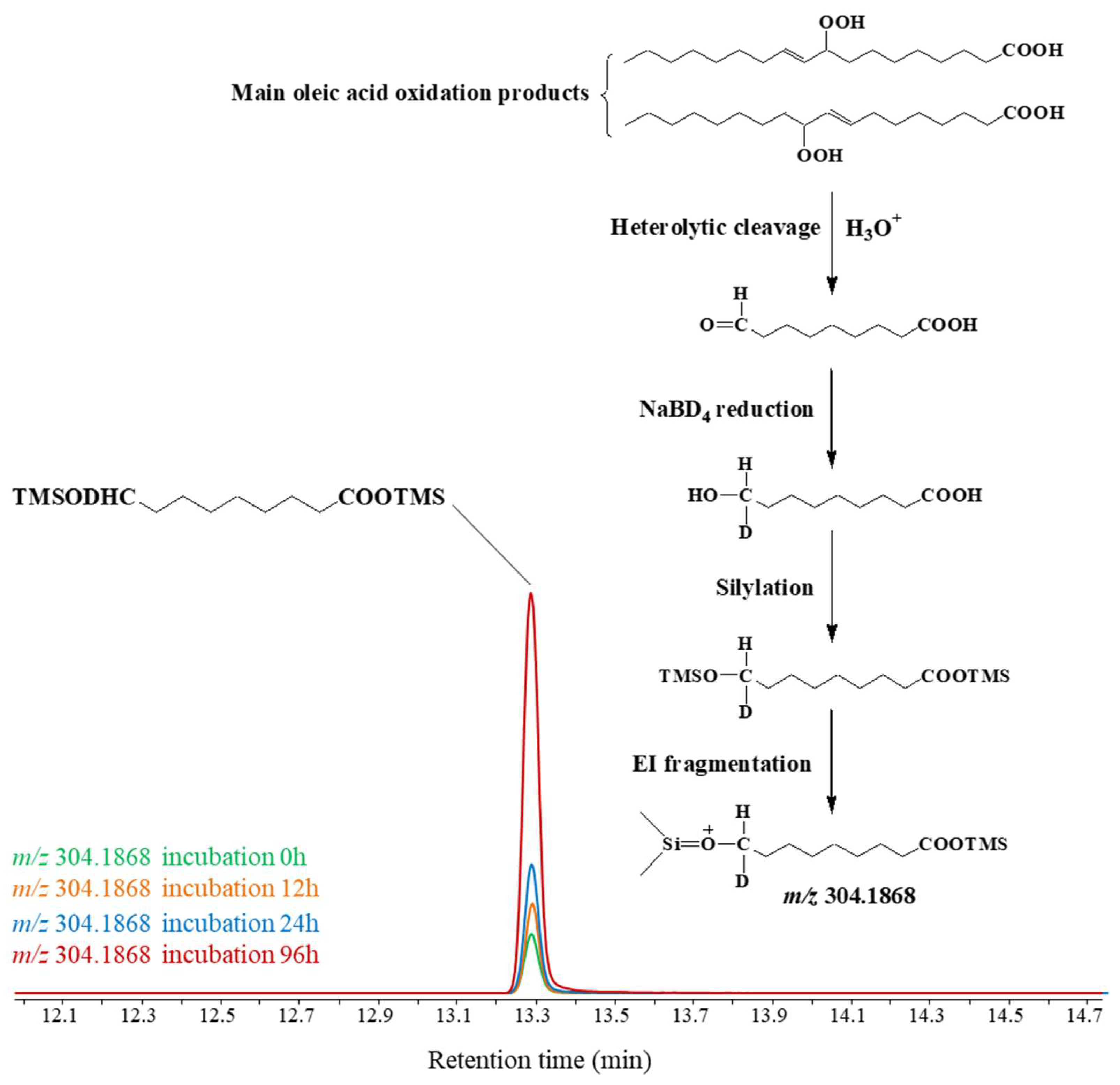
The involvement of such degradative processes was confirmed by the increasing amounts of deuterated ω-hydroxynonanoic acid detected after NaBD4 reduction of dust samples incubated in fresh water and that signal heterolytic cleavage of oleic acid oxidation products to ω-oxononanoic acid during the incubation (Figure 6). Note that 11-oxoamyrons also can be degraded after 1,4-addition of water on their conjugated 11-oxo group and retro-aldol reaction [71]. The very fast degradation of the stable hydroperoxides resulting from amyrin oxidation observed at the pH of the fresh water employed (6.8), which is in the high range of pH generally found in rainwater [18,72], strongly suggests that wet atmospheric inputs from the Negev desert arriving at sea would be very poor in hydroperoxides.
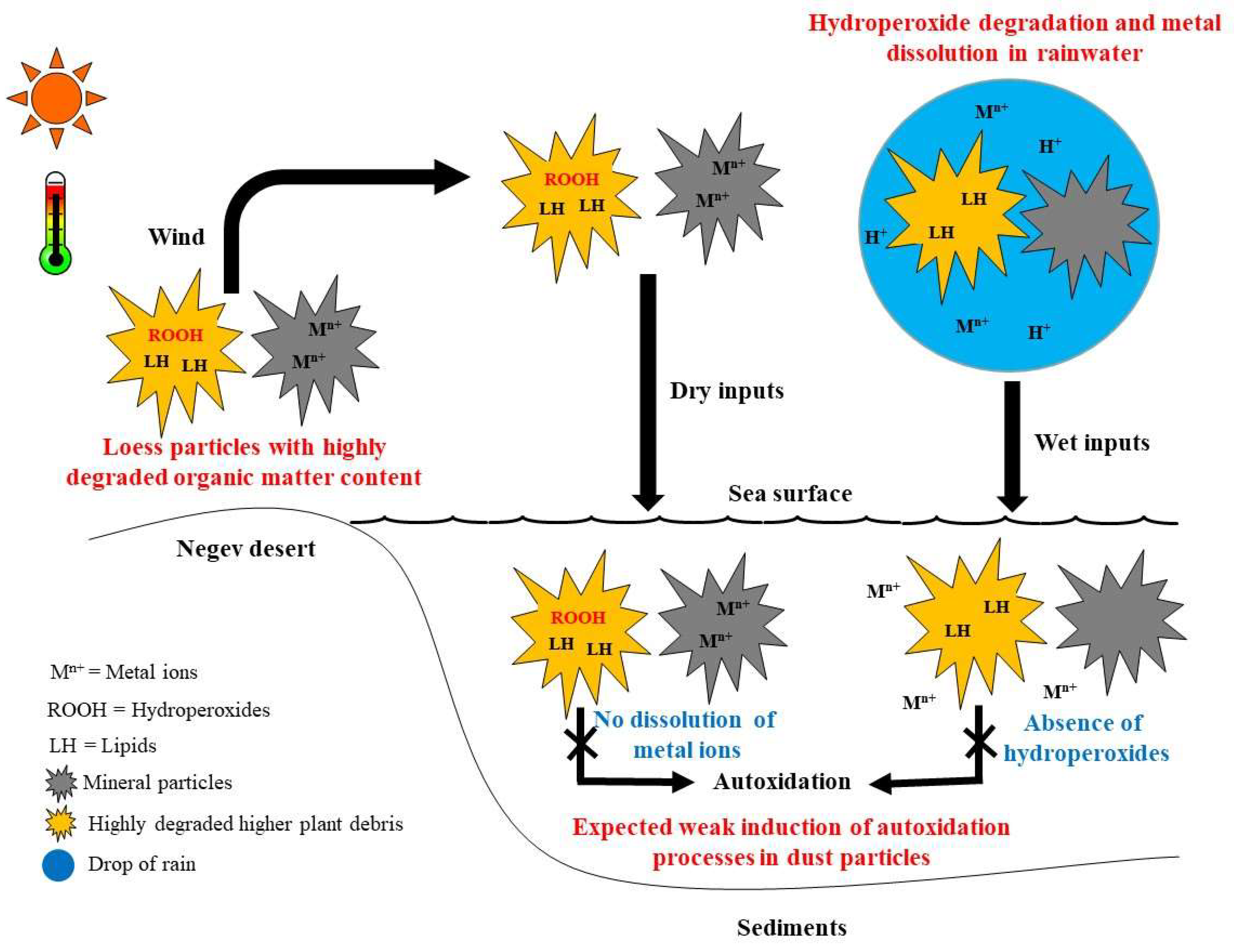
Autoxidation processes are generally induced by homolytic cleavage of peroxy bonds by redox-active metal ions undergoing one-electron transfer, UV radiation, high temperatures, or lipoxygenases [9,19]. Despite the release of some redox-active metal components of dust analogue in rainwater (see above), their very low content of hydroperoxides resulting from (i) homolytic cleavage under the effect of high desert-environment temperatures and solar irradiances and (ii) heterolytic cleavage in rainwater would substantially limit autoxidative alterations of this material at sea (Figure 7). If the small quantities of hydroperoxides present in the dry inputs can reach the sea without being degraded, they should be preserved there, as the relatively high pH of the seawater limits the dissolution of metal ions [18] (Figure 7). Given the moderate temperatures of Mediterranean surface seawater (ranging from 13 °C to 26 °C according to season [73]) and the well-known rapid attenuation of solar UV radiation in seawater [74], it seems very unlikely that these parameters would have an effect on the induction of autoxidation processes in atmospheric dust inputs. As previously demonstrated in river estuaries [9], residual hydroperoxides also can be cleaved by the action of lipoxygenases present in plant debris and whose activity intensifies with increasing salinity. However, this hypothesis can be rejected here, due to the strongly detrital nature of the Negev loess deposits and the total absence of PUFAs (lipoxygenase substrates [75]) found in them.
4. Conclusions
Analyses of lipids and their oxidation products in loess material from the Negev Desert demonstrated that higher plant components undergo intense autoxidation in the desert environment, mainly affording hydroperoxides. Due to the extreme environmental conditions present in this desert (high temperatures and solar irradiance), any lipid oxidation products quickly get degraded and only the more stable products (11-hydroperoxyamyrins) accumulate. Incubation of these loess samples in fresh water (in order to mimic their behavior in rainwater) showed intense dissolution of some metals (mainly iron) and fast heterolytic degradation of the residual hydroperoxides. During the deposition of dry atmospheric dusts originating from the Negev desert into the Mediterranean Sea, the autoxidation of higher plant components (which cannot be induced without the simultaneous presence of metal ions and hydroperoxides) should be very weak due to the limited capacity for metal dissolution at the typical pH of seawater. If metal dissolution is strongly favored in wet atmospheric inputs (due to their low pH), then there should also be intense heterolysis of hydroperoxides under these conditions, which would again limit the induction of autoxidation processes. As a consequence, autoxidative degradation of lipid components of dust particles arising from the Negev desert should be very weak in seawater. The low intensity of these processes should therefore favor the preservation of plant material transported by atmospheric inputs in marine sediments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/w14244092/s1, Table S1: Concentrations of the different lipids detected in Negev loess deposits (results of triplicates); Table S2: Concentrations (µg/g) of lipids and their oxidation products in Negev loess deposits (means of triplicates); Table S3: Concentrations of α- and β-amyrin oxidation products before and after incubation in fresh water.
Author Contributions
Conceptualization, J.-F.R., B.C. and D.A.; Methodology, J.-F.R., B.C. and C.M.; Formal analysis, J.-F.R., B.C. and C.M.; Sampling, I.K.; Writing—Original draft preparation, J.-F.R., B.C. and D.A.; Writing—Review and editing, J.-F.R., B.C. and D.A.; Project administration, J.-F.R.; Funding acquisition, J.-F.R., B.C. and D.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the LEFE-CYBER (Les Enveloppes Fluides et l’Environnement) national program, as part of the ATMOSMER (Etude de la dégradation abiotique des apports ATMOSphériques Sahariens en MER et de leur impact sur la dégradation des communautés phytoplanctoniques marines) research program. ERDF (European Regional Development Fund) (Oceanomed project, No. 1166-39417) provided funding for the apparatuses employed.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Leeuw, J.W.; Largeau, C. A Review of Macromolecular Organic Compounds That Comprise Living Organisms and Their Role in Kerogen, Coal, and Petroleum Formation. In Organic Geochemistry; Engel, M.H., Macko, S.A., Eds.; Topics in Geobiology; Springer US: Boston, MA, USA, 1993; Volume 11, pp. 23–72. ISBN 978-1-4613-6252-4. [Google Scholar]
- Wakeham, S.G.; Canuel, E.A. Degradation and Preservation of Organic Matter in Marine Sediments. In Marine Organic Matter: Biomarkers, Isotopes and DNA; Volkman, J.K., Ed.; The Handbook of Environmental Chemistry; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 2N, pp. 295–321. ISBN 978-3-540-28401-7. [Google Scholar]
- Hedges, J.I.; Keil, R.G. Sedimentary Organic Matter Preservation: An Assessment and Speculative Synthesis. Mar. Chem. 1995, 49, 81–115. [Google Scholar] [CrossRef]
- Vonk, J.E.; Sánchez-García, L.; Semiletov, I.; Dudarev, O.; Eglinton, T.; Andersson, A.; Gustafsson, Ö. Molecular and Radiocarbon Constraints on Sources and Degradation of Terrestrial Organic Carbon along the Kolyma Paleoriver Transect, East Siberian Sea. Biogeosciences 2010, 7, 3153–3166. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.S.; Charkin, A.; Dudarev, O.; Semiletov, I.; Vonk, J.E.; Sánchez-García, L.; Andersson, A.; Gustafsson, Ö. Carbon Isotopes and Lipid Biomarker Investigation of Sources, Transport and Degradation of Terrestrial Organic Matter in the Buor-Khaya Bay, SE Laptev Sea. Biogeosciences 2011, 8, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Rontani, J.-F. Photo- and Free Radical-Mediated Oxidation of Lipid Components During the Senescence of Phototrophic Organisms. In Senescence; Nagata, T., Ed.; InTech: Saitama, Japan, 2012; ISBN 978-953-51-0144-4. [Google Scholar]
- Rontani, J.-F.; Charrière, B.; Sempéré, R.; Doxaran, D.; Vaultier, F.; Vonk, J.E.; Volkman, J.K. Degradation of Sterols and Terrigenous Organic Matter in Waters of the Mackenzie Shelf, Canadian Arctic. Org. Geochem. 2014, 75, 61–73. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Galeron, M.-A.; Amiraux, R.; Artigue, L.; Belt, S.T. Identification of Di- and Triterpenoid Lipid Tracers Confirms the Significant Role of Autoxidation in the Degradation of Terrestrial Vascular Plant Material in the Canadian Arctic. Org. Geochem. 2017, 108, 43–50. [Google Scholar] [CrossRef]
- Galeron, M.-A.; Radakovitch, O.; Charrière, B.; Vaultier, F.; Volkman, J.K.; Bianchi, T.S.; Ward, N.D.; Medeiros, P.M.; Sawakuchi, H.O.; Tank, S.; et al. Lipoxygenase-Induced Autoxidative Degradation of Terrestrial Particulate Organic Matter in Estuaries: A Widespread Process Enhanced at High and Low Latitude. Org. Geochem. 2018, 115, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Bonin, P.; Prime, A.; Galeron, M.; Guasco, S.; Rontani, J. Enhanced Biotic Degradation of Terrestrial POM in an Estuarine Salinity Gradient: Interactive Effects of Organic Matter Pools and Changes of Bacterial Communities. Aquat. Microb. Ecol. 2019, 83, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Rontani, J.-F.; Belt, S.T. Photo- and Autoxidation of Unsaturated Algal Lipids in the Marine Environment: An Overview of Processes, Their Potential Tracers, and Limitations. Org. Geochem. 2020, 139, 103941. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Chemical Modification of Proteins by Reactive Oxygen Species. In Redox Proteomics; Dalle-Donne, I., Scaloni, A., Butterfield, D.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 1–23. ISBN 978-0-471-97312-6. [Google Scholar]
- Barreca, A.M.; Fabbrini, M.; Galli, C.; Gentili, P.; Ljunggren, S. Laccase/Mediated Oxidation of a Lignin Model for Improved Delignification Procedures. J. Mol. Catal. B Enzym. 2003, 26, 105–110. [Google Scholar] [CrossRef]
- Bianchi, T.S. The Role of Terrestrially Derived Organic Carbon in the Coastal Ocean: A Changing Paradigm and the Priming Effect. Proc. Natl. Acad. Sci. USA 2011, 108, 19473–19481. [Google Scholar] [CrossRef]
- d’Almeida, G.A. A Model for Saharan Dust Transport. J. Clim. Appl. Meteor. 1986, 25, 903–916. [Google Scholar] [CrossRef]
- Marticorena, B.; Bergametti, G.; Aumont, B.; Callot, Y.; N’Doumé, C.; Legrand, M. Modeling the Atmospheric Dust Cycle: 2. Simulation of Saharan Dust Sources. J. Geophys. Res. 1997, 102, 4387–4404. [Google Scholar] [CrossRef]
- Schefuß, E.; Ratmeyer, V.; Stuut, J.-B.W.; Jansen, J.H.F.; Damsté, J.S.S. Carbon Isotope Analyses of N-Alkanes in Dust from the Lower Atmosphere over the Central Eastern Atlantic. Geochim. Et Cosmochim. Acta 2003, 67, 1757–1767. [Google Scholar] [CrossRef]
- Korte, L.F.; Pausch, F.; Trimborn, S.; Brussaard, C.P.D.; Brummer, G.-J.A.; van der Does, M.; Guerreiro, C.V.; Schreuder, L.T.; Munday, C.I.; Stuut, J.-B.W. Effects of Dry and Wet Saharan Dust Deposition in the Tropical North Atlantic Ocean. Biogeosci. Discuss. 2018, 1–20. [Google Scholar]
- Schaich, K.M. Lipid Oxidation: Theoretical Aspects. In Bailey’s Industrial Oil and Fat Products; Shahidi, F., Ed.; Wiley: Hoboken, NJ, USA, 2005; ISBN 978-0-471-38460-1. [Google Scholar]
- Kouyoumdjian, H.; Saliba, N.A. Ion Concentrations of PM 10-2.5 and PM 2.5 Aerosols Over the Eastern Mediterranean Region: Seasonal Variation and Source Identification. Atmos. Chem. Phys. Discuss. 2005, 5, 13053–13073. [Google Scholar]
- Swet, N.; Katra, I. Reduction in Soil Aggregation in Response to Dust Emission Processes. Geomorphology 2016, 268, 177–183. [Google Scholar] [CrossRef]
- Katra, I.; Gross, A.; Swet, N.; Tanner, S.; Krasnov, H.; Angert, A. Substantial Dust Loss of Bioavailable Phosphorus from Agricultural Soils. Sci. Rep. 2016, 6, 24736. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Katra, I.; Schaeur, J.J.; Rudich, Y. Mitochondria-mediated Oxidative Stress Induced by Desert Dust in Rat Alveolar Macrophages. GeoHealth 2017, 1, 4–16. [Google Scholar] [CrossRef]
- Elad, D.; Zaretsky, U.; Avraham, S.; Gotlieb, R.; Wolf, M.; Katra, I.; Sarig, S.; Zaady, E. In Vitro Exposure of Nasal Epithelial Cells to Atmospheric Dust. Biomech. Model. Mechanobiol. 2018, 17, 891–901. [Google Scholar] [CrossRef]
- Zhan, Y.; Ginder-Vogel, M.; Shafer, M.M.; Rudich, Y.; Pardo, M.; Katra, I.; Katoshevski, D.; Schauer, J.J. Changes in Oxidative Potential of Soil and Fly Ash after Reaction with Gaseous Nitric Acid. Atmos. Environ. 2018, 173, 306–315. [Google Scholar] [CrossRef]
- Gross, A.; Tiwari, S.; Shtein, I.; Erel, R. Direct Foliar Uptake of Phosphorus from Desert Dust. New Phytol. 2021, 230, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y. Integrated Wind-Erosion Modelling. In Physics and Modelling of Wind Erosion; Shao, Y., Ed.; Atmospheric and Oceanographic Sciences Library; Springer Netherlands: Dordrecht, The Netherlands, 2009; Volume 37, pp. 303–360. ISBN 978-1-4020-8894-0. [Google Scholar]
- Herut, B.; Nimmo, M.; Medway, A.; Chester, R.; Krom, M.D. Dry Atmospheric Inputs of Trace Metals at the Mediterranean Coast of Israel (SE Mediterranean): Sources and Fluxes. Atmos. Environ. 2001, 35, 803–813. [Google Scholar] [CrossRef]
- Ganor, E.; Foner, H.A. Mineral Dust Concentrations, Deposition Fluxes and Deposition Velocities in Dust Episodes over Israel. J. Geophys. Res. 2001, 106, 18431–18437. [Google Scholar] [CrossRef] [Green Version]
- Katra, I.; Arotsker, L.; Krasnov, H.; Zaritsky, A.; Kushmaro, A.; Ben-Dov, E. Richness and Diversity in Dust Stormborne Biomes at the Southeast Mediterranean. Sci. Rep. 2015, 4, 5265. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Palchan, D.; Krom, M.D.; Angert, A. Elemental and Isotopic Composition of Surface Soils from Key Saharan Dust Sources. Chem. Geol. 2016, 442, 54–61. [Google Scholar] [CrossRef]
- Uni, D.; Katra, I. Airborne Dust Absorption by Semi-Arid Forests Reduces PM Pollution in Nearby Urban Environments. Sci. Total Environ. 2017, 598, 984–992. [Google Scholar] [CrossRef]
- Dumas, C.; Aubert, D.; de Madron, X.D.; Ludwig, W.; Heussner, S.; Delsaut, N.; Menniti, C.; Sotin, C.; Buscail, R. Storm-Induced Transfer of Particulate Trace Metals to the Deep-Sea in the Gulf of Lion (NW Mediterranean Sea). Environ. Geochem. Health 2014, 36, 995–1014. [Google Scholar] [CrossRef]
- Maring, H.B.; Duce, R.A. The Impact of Atmospheric Aerosols on Trace Metal Chemistry in Open Ocean Surface Seawater, 1. Aluminum. Earth Planet. Sci. Lett. 1987, 84, 381–392. [Google Scholar] [CrossRef]
- Chester, R.; Nimmo, M.; Corcoran, P.A. Rain Water-Aerosol Trace Metal Relationships at Cap Ferrat: A Coastal Site in the Western Mediterranean. Mar. Chem. 1997, 58, 293–312. [Google Scholar] [CrossRef]
- Desboeufs, K.V.; Losno, R.; Vimeux, F.; Cholbi, S. The PH-Dependent Dissolution of Wind-Transported Saharan Dust. J. Geophys. Res. 1999, 104, 21287–21299. [Google Scholar] [CrossRef]
- Longo, A.F.; Feng, Y.; Lai, B.; Landing, W.M.; Shelley, R.U.; Nenes, A.; Mihalopoulos, N.; Violaki, K.; Ingall, E.D. Influence of Atmospheric Processes on the Solubility and Composition of Iron in Saharan Dust. Environ. Sci. Technol. 2016, 50, 6912–6920. [Google Scholar] [CrossRef] [PubMed]
- Meskhidze, N. Dust and Pollution: A Recipe for Enhanced Ocean Fertilization? J. Geophys. Res. 2005, 110, D03301. [Google Scholar] [CrossRef]
- Hettiarachchi, E.; Reynolds, R.L.; Goldstein, H.L.; Moskowitz, B.; Rubasinghege, G. Bioavailable Iron Production in Airborne Mineral Dust: Controls by Chemical Composition and Solar Flux. Atmos. Environ. 2019, 205, 90–102. [Google Scholar] [CrossRef]
- Desboeufs, K.V.; Sofikitis, A.; Losno, R.; Colin, J.L.; Ausset, P. Dissolution and Solubility of Trace Metals from Natural and Anthropogenic Aerosol Particulate Matter. Chemosphere 2005, 58, 195–203. [Google Scholar] [CrossRef]
- Herut, B.; Starinsky, A.; Katz, A.; Rosenfeld, D. Relationship between the Acidity and Chemical Composition of Rainwater and Climatological Conditions along a Transition Zone between Large Deserts and Mediterranean Climate, Israel. Atmos. Environ. 2000, 34, 1281–1292. [Google Scholar] [CrossRef]
- Ridame, C.; Guieu, C. Saharan Input of Phosphate to the Oligotrophic Water of the Open Western Mediterranean Sea. Limnol. Oceanogr. 2002, 47, 856–869. [Google Scholar] [CrossRef]
- Rontani, J.F.; Charrière, B.; Menniti, C.; Aubert, D.; Aubert, C. Electron Ionization Mass Spectrometry Fragmentation and Multiple Reaction Monitoring Quantification of Autoxidation Products of α- and β-Amyrins in Natural Samples. Rapid Commun. Mass Spectrom. 2018, 32, 1599–1607. [Google Scholar] [CrossRef]
- Mihara, S.; Tateba, H. Photosensitized Oxygenation Reactions of Phytol and Its Derivatives. J. Org. Chem. 1986, 51, 1142–1144. [Google Scholar] [CrossRef]
- Rontani, J.-F. Lipid Oxidation Products: Useful Tools for Monitoring Photo- and Autoxidation in Phototrophs; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2021; ISBN 978-1-5275-7359-8. [Google Scholar]
- Shalom, O.; Crouvi, O.; Enzel, Y.; Rosenfeld, D. Locally Recycled Late Pleistocene Loess Feeds Modern Dust Storms at the Desert Margins of the Eastern Mediterranean, Israel. Aeolian Res. 2020, 46, 100612. [Google Scholar] [CrossRef]
- Sicre, M.-A.; Paillasseur, J.-L.; Marty, J.-C.; Saliot, A. Characterization of Seawater Samples Using Chemometric Methods Applied to Biomarker Fatty Acids. Org. Geochem. 1988, 12, 281–288. [Google Scholar] [CrossRef]
- McGrath, C.F.; Moss, C.W.; Burchard, R.P. Effect of Temperature Shifts on Gliding Motility, Adhesion, and Fatty Acid Composition of Cytophaga Sp. Strain U67. J. Bacteriol. 1990, 172, 1978–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, A.H.B.; Holloway, P.J. The Intermolecular Structure of Some Plant Cutins. In Lipids and Lipid Polymers in Higher Plants; Tevini, M., Lichtenthaler, H.K., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 1977; pp. 293–299. ISBN 978-3-642-66634-6. [Google Scholar]
- Kolattukudy, P.E. Biopolyester Membranes of Plants: Cutin and Suberin. Science 1980, 208, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Bichot, A.; Delgenes, J.P.; Radoiu, M.; Garcia Bernet, D. Microwave Pretreatment of Lignocellulosic Biomass to Release Maximum Phenolic Acids. In Proceedings of the 17th International Conference on Microwave and High Frequency Heating, Barcelona, Spain, 9 September 2019. [Google Scholar]
- Volkman, J. Sterols in Microorganisms. Appl. Microbiol. Biotechnol. 2003, 60, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Aziz, M.; Sturtevant, D.; Chapman, K.D.; Guo, L. Heterogeneous Distribution of Erucic Acid in Brassica Napus Seeds. Front. Plant Sci. 2020, 10, 1744. [Google Scholar] [CrossRef] [Green Version]
- Brassell, S.C.; Eglinton, G.; Maxwell, J.R. The Geochemistry of Terpenoids and Steroids. Biochem. Soc. Trans. 1983, 11, 575–586. [Google Scholar] [CrossRef]
- Otto, A.; Simpson, M.J. Degradation and Preservation of Vascular Plant-Derived Biomarkers in Grassland and Forest Soils from Western Canada. Biogeochemistry 2005, 74, 377–409. [Google Scholar] [CrossRef]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.; Scheffler, A. Pentacyclic Triterpene Distribution in Various Plants—Rich Sources for a New Group of Multi-Potent Plant Extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [Green Version]
- Knoche, H.W. A Study on the Biosynthesis Ofcis-9,10-Epoxyoctadecanoic Acid. Lipids 1968, 3, 163–169. [Google Scholar] [CrossRef]
- Marchand, D.; Rontani, J.-F. Characterisation of Photo-Oxidation and Autoxidation Products of Phytoplanktonic Monounsaturated Fatty Acids in Marine Particulate Matter and Recent Sediments. Org. Geochem. 2001, 32, 287–304. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Aubert, C.; Belt, S.T. EIMS Fragmentation Pathways and MRM Quantification of 7α/β-Hydroxy-Dehydroabietic Acid TMS Derivatives. J. Am. Soc. Mass Spectrom. 2015, 26, 1606–1616. [Google Scholar] [CrossRef]
- Galeron, M.-A.; Vaultier, F.; Rontani, J.-F. Oxidation Products of α- and β-Amyrins: Potential Tracers of Abiotic Degradation of Vascular-Plant Organic Matter in Aquatic Environments. Environ. Chem. 2016, 13, 732. [Google Scholar] [CrossRef]
- Buanafina, M.M.D.O. Feruloylation in Grasses: Current and Future Perspectives. Mol. Plant 2009, 2, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Minatel, I.O.; Borges, C.V.; Ferreira, M.I.; Gomez, H.A.G.; Chen, C.-Y.O.; Lima, G.P.P. Phenolic Compounds: Functional Properties, Impact of Processing and Bioavailability. In Phenolic Compounds—Biological Activity; Soto-Hernndez, M., Palma-Tenango, M., Garcia-Mateos, M.D.R., Eds.; InTech: Saitama, Japan, 2017; ISBN 978-953-51-2959-2. [Google Scholar]
- Katra, I. Soil Erosion by Wind and Dust Emission in Semi-Arid Soils Due to Agricultural Activities. Agronomy 2020, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Crouvi, O.; Amit, R.; Ben Israel, M.; Enzel, Y. Loess in the Negev Desert: Sources, Loessial Soils, Palaeosols, and Palaeoclimatic Implications. In Quaternary of the Levant; Enzel, Y., Bar-Yosef, O., Eds.; Cambridge University Press: Cambridge, UK, 2017; pp. 471–482. ISBN 978-1-316-10675-4. [Google Scholar]
- Wedepohl, K.H. The Composition of the Continental Crust. Geochim. Et Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Lucke, B.; Sandler, A.; Vanselow, K.A.; Bruins, H.J.; Abu-Jaber, N.; Bäumler, R.; Porat, N.; Kouki, P. Composition of Modern Dust and Holocene Aeolian Sediments in Archaeological Structures of the Southern Levant. Atmosphere 2019, 10, 762. [Google Scholar] [CrossRef] [Green Version]
- Mahowald, N.M.; Hamilton, D.S.; Mackey, K.R.M.; Moore, J.K.; Baker, A.R.; Scanza, R.A.; Zhang, Y. Aerosol Trace Metal Leaching and Impacts on Marine Microorganisms. Nat. Commun. 2018, 9, 2614. [Google Scholar] [CrossRef] [Green Version]
- Frimer, A.A. The Reaction of Singlet Oxygen with Olefins: The Question of Mechanism. Chem. Rev. 1979, 79, 359–387. [Google Scholar] [CrossRef]
- Turner, J.A.; Herz, W. Iron(II)-Induced Decomposition of Unsaturated Cyclic Peroxides Derived from Butadienes. A Simple Procedure for Synthesis of 3-Alkylfurans. J. Org. Chem. 1977, 42, 1900–1904. [Google Scholar] [CrossRef]
- Rontani, J.-F. Photodegradation of Unsaturated Fatty Acids in Senescent Cells of Phytoplankton: Photoproduct Structural Identification and Mechanistic Aspects. J. Photochem. Photobiol. A Chem. 1998, 114, 37–44. [Google Scholar] [CrossRef]
- Vashchenko, E.V.; Knyazeva, I.V.; Krivoshey, A.I.; Vashchenko, V.V. Retro-Aldol Reactions in Micellar Media. Mon. Chem. 2012, 143, 1545–1549. [Google Scholar] [CrossRef]
- Shi, C.; Deng, X.; Yang, Y.; Huang, X.; Wu, B. Precipitation Chemistry and Corresponding Transport Patterns of Influencing Air Masses at Huangshan Mountain in East China. Adv. Atmos. Sci. 2014, 31, 1157–1166. [Google Scholar] [CrossRef]
- Shaltout, M.; Omstedt, A. Recent sea surface temperature trends and future scenarios for the Mediterranean Sea. Oceanologia 2014, 56, 411–443. [Google Scholar] [CrossRef]
- Brown, B.E.; Dunne, R.P.; Chansang, H. Coral Bleaching Relative to Elevated Seawater Temperature in the Andaman Sea (Indian Ocean) over the Last 50 Years. Coral Reefs 1996, 15, 151–152. [Google Scholar] [CrossRef]
- Garreta, A.; Val-Moraes, S.P.; García-Fernández, Q.; Busquets, M.; Juan, C.; Oliver, A.; Ortiz, A.; Gaffney, B.J.; Fita, I.; Manresa, À.; et al. Structure and Interaction with Phospholipids of a Prokaryotic Lipoxygenase from Pseudomonas Aeruginosa. FASEB J. 2013, 27, 4811–4821. [Google Scholar] [CrossRef]
Figure 1.
Oxidation state of the main lipids detected in Negev loess deposits.

Figure 2.
Concentration of trace metals during incubation experiment in mQ water.

Figure 3.
Behaviour of amyrin oxidation products during the different treatments.

Figure 4.
GC-QTOF characterization of amyrin oxidation products after deuteration.

Figure 5.
Time-course degradation of amyrin oxidation products in fresh water.

Figure 6.
TOF chromatograms showing the production of increasing amounts of deuterated ω-hydroxynonanoic acid (arising from heterolytic cleavage of oleic oxidation products) during incubation of dust analogue in fresh water.
Figure 6.
TOF chromatograms showing the production of increasing amounts of deuterated ω-hydroxynonanoic acid (arising from heterolytic cleavage of oleic oxidation products) during incubation of dust analogue in fresh water.

Figure 7.
Conceptual scheme showing the behaviour of hydroperoxides and metal ions in dry and wet atmospheric dust inputs and in seawater.
Figure 7.
Conceptual scheme showing the behaviour of hydroperoxides and metal ions in dry and wet atmospheric dust inputs and in seawater.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Concentrations and origins of the lipid components of Negev loess deposits.
| Lipid | Concentration | Origin |
|---|---|---|
| C18:1Δ9 (oleic) acid | 43.0 ± 10.0 a | Plants, fungi, bacteria |
| C18:1Δ11 (vaccenic) acid | 5.1 ± 1.3 a | Bacteria |
| C18:2Δ9,12 (linoleic) acid | 8.2 ± 0.9 a | Plants, fungi |
| C22:1Δ13 (erucic) acid | 6.4 ± 1.3 a | Brassicaceae |
| ω,(9-10)-dihydroxy-C16:0 acids 9,10-epoxyoctadecanoic acid c Dehydroabietic acid | 22.3 ± 2.7 a 10.2 ± 1.9 a 4.1 ± 1.9 a | Higher plants (cuticular waxes) Fungi-infected plants Gymnosperms |
| p-coumaric acid | 0.6 ± 0.1 a | Higher plants (lignocellulose) |
| Ferulic acid | 2.4 ± 0.3 a | Higher plants (lignocellulose) |
| Cholesterol | 8.8 ± 3.1 a | Plants, fungi, animals |
| Campesterol | 0.5 ± 0.1 a | Higher plants |
| Sitosterol Ergosterol | 0.6 ± 0.1 a 0.3 ± 0.1 a | Higher plants Fungi, yeasts |
| α-amyrin | 24.2 ± 8.2 b | Angiosperms |
| β-amyrin | 26.6 ± 3.7 b | Angiosperms, fungi |
a µg g−1, b ng g−1, c detected in the form of the corresponding methoxyhydrins, 9,10-diols and chlorohydrins.
Table 2.
Total metal concentrations (µg g−1) in dust analogue collected in northern Negev and percent release relative to initial content in the particulate phase during incubation experiments (T0, T12h, T24h and T96h) and the respective SD.
Table 2.
Total metal concentrations (µg g−1) in dust analogue collected in northern Negev and percent release relative to initial content in the particulate phase during incubation experiments (T0, T12h, T24h and T96h) and the respective SD.
| Concentrations | Dust Sample | T0 | T12h | T24h | T96h |
|---|---|---|---|---|---|
| Ca | 137,988 ± 13,990 | 2.21 ± 0.13 | 4.02 ± 0.15 | 4.42 ± 0.10 | 4.11 ± 0.24 |
| Fe | 32,589 ± 3143 | 0.06 ± 0.01 | 0.68 ± 0.25 | 1.55 ± 0.51 | 0.02 ± 0.01 |
| Al | 41,357 ± 3580 | 0.10 ± 0.01 | 0.90 ± 0.21 | 1.84 ± 0.61 | 0.27 ± 0.01 |
| Zn | 77.1 ± 1.3 | 15.62 ± 3.77 | 10.86 ± 11.50 | 13.09 ± 11.85 | 2.73 ± 1.07 |
| Cu | 21.4 ± 1.9 | 3.43 ± 0.96 | 2.58 ± 0.69 | 3.44 ± 0.26 | 4.37 ± 2.87 |
| V | 112 ± 11 | 0.56 ± 0.02 | 2.12 ± 0.21 | 3.50 ± 0.32 | 2.73 ± 0.05 |
| Mn | 613 ± 63 | 0.21 ± 0.034 | 0.68 ± 0.29 | 1.29 ± 0.35 | 0.06 ± 0.02 |
| Co | 13.7 ± 1.5 | 0.17 ± 0.031 | 1.03 ± 0.35 | 1.91 ± 0.54 | 0.31 ± 0.06 |
| Cd | 0.63 ± 0.09 | 2.59 ± 1.11 | 2.01 ± 0.68 | 2.89 ± 1.08 | 1.13 ± 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rontani, J.-F.; Charriere, B.; Menniti, C.; Katra, I.; Aubert, D. Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components. Water 2022, 14, 4092. https://doi.org/10.3390/w14244092
AMA Style
Rontani J-F, Charriere B, Menniti C, Katra I, Aubert D. Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components. Water. 2022; 14(24):4092. https://doi.org/10.3390/w14244092
Chicago/Turabian StyleRontani, Jean-François, Bruno Charriere, Christophe Menniti, Itzhak Katra, and Dominique Aubert. 2022. "Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components" Water 14, no. 24: 4092. https://doi.org/10.3390/w14244092
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.