
Comparison of the Efficiency of Single-Locus Species Delimitation Methods: A Case Study of a Single Lake Fish Population in Comparison against the Barcodes from International Databases

 ,
,  ,
,
Abstract
:Epigraph:“What’s the use of their having names,” the Gnat said, “if they won’t answer to them?”“No use to them,’ said Alice; ‘but it’s useful to the people who name them, I suppose. If not, why do things have names at all?”Lewis Carroll “Through the Looking-Glass and what Alice found there” (1871).
1. Introduction
2. Materials and Methods
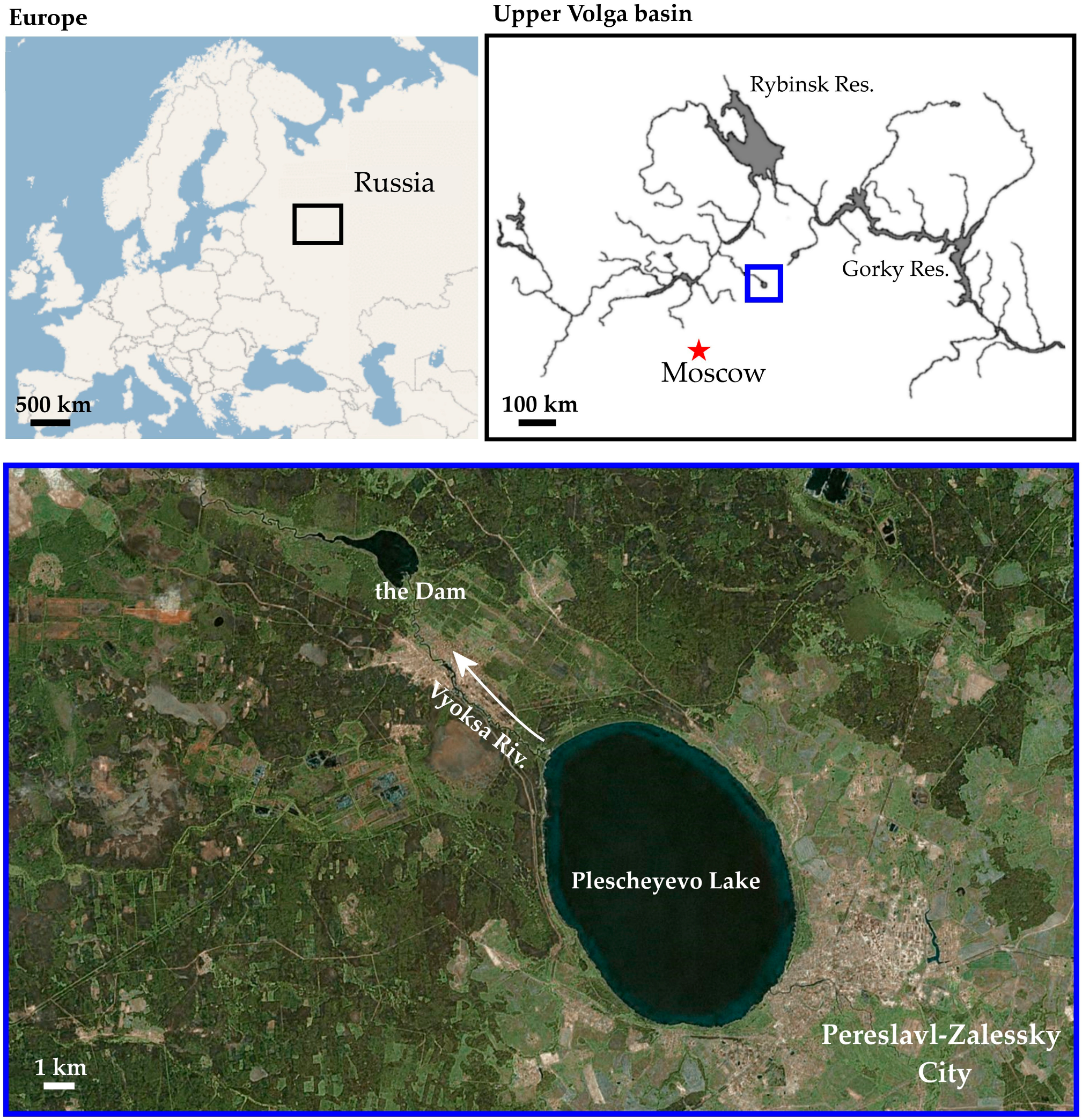
2.1. Sampling
2.2. From DNA Extraction to Phylogenetic Analysis
2.3. Different Methods of mOTUs Delimitation
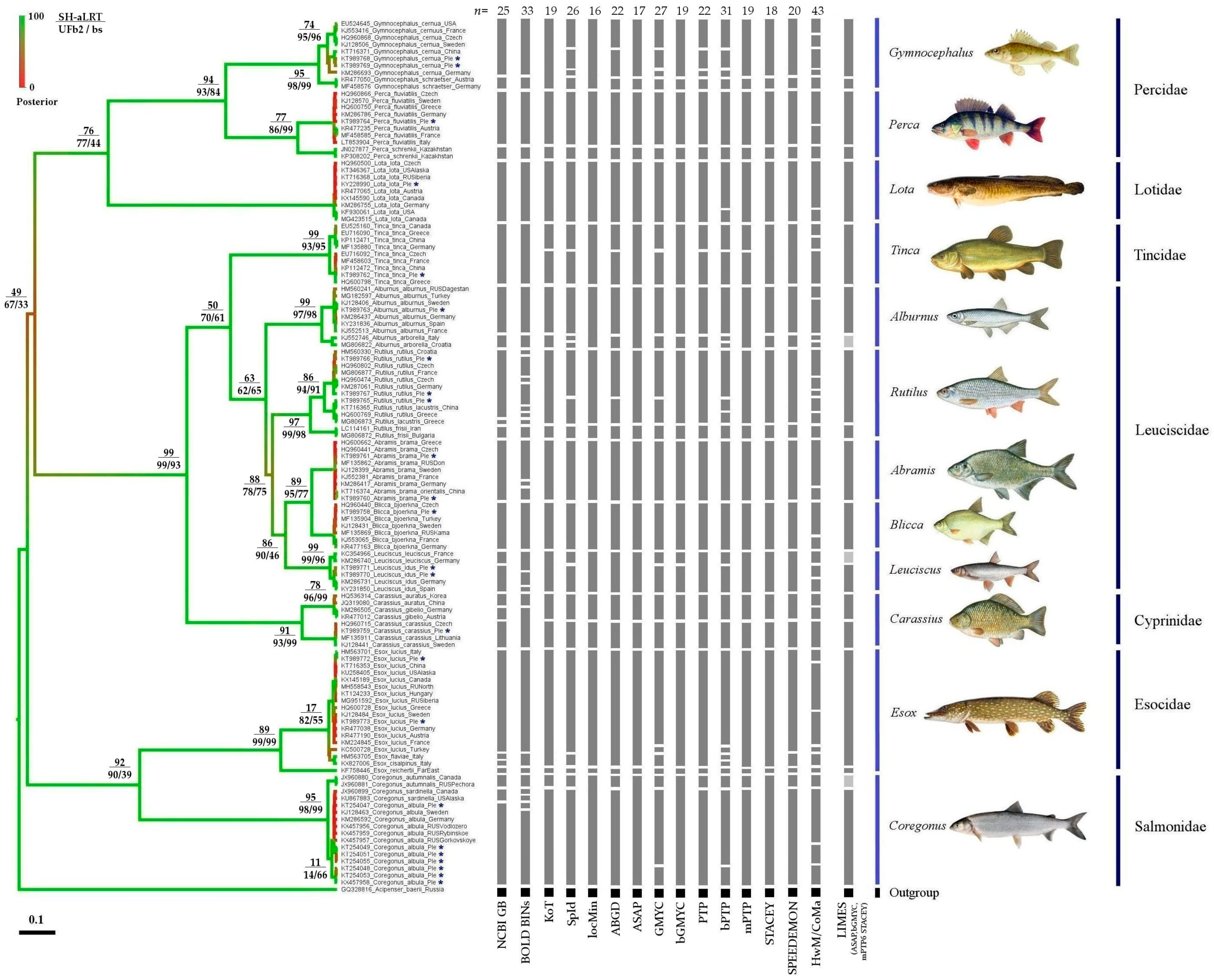
3. Results
3.1. Phylogenetic Analysis
3.2. Comparison of Different Approaches to mOTU Delimitation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- UNCED. AGENDA 21: The Earth Summit Strategy to Save Our Planet. Available online: https://sustainabledevelopment.un.org/outcomedocuments/agenda21 (accessed on 7 July 2022).
- Templeton, A.R. Using phylogeographic analyses of gene trees to test species status and processes. Mol. Ecol. 2001, 10, 779–791. [Google Scholar] [CrossRef] [PubMed]
- van Klink, R.; Bowler, D.E.; Gongalsky, K.B.; Swengel, A.B.; Gentile, A.; Chase, J.M. Meta-analysis reveals declines in terrestrial but increases in freshwater insect abundances. Science 2020, 368, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Trewick, S.A. DNA Barcoding is not enough: Mismatch of taxonomy and genealogy in New Zealand grasshoppers (Orthoptera: Acrididae). Cladistics 2008, 24, 240–254. [Google Scholar] [CrossRef]
- Ebach, M.C.; de Carvalho, M.R. Anti-intellectualism in the DNA barcoding enterprise. Zoologia 2010, 27, 165–178. [Google Scholar] [CrossRef]
- Taylor, H.R.; Harris, W.E. An emergent science on the brink of irrelevance: A review of the past 8 years of DNA barcoding. Mol. Ecol. Resour. 2012, 12, 377–388. [Google Scholar] [CrossRef]
- Garibian, P.G.; Neretina, A.N.; Taylor, D.J.; Kotov, A.A. Partial revision of the neustonic genus Scapholeberis Schoedler, 1858 (Crustacea: Cladocera): Decoding of the barcoding results. PeerJ 2020, 8, e10410. [Google Scholar] [CrossRef]
- Zamani, A.; Fric, Z.F.; Gante, H.F.; Hopkins, T.; Orfinger, A.B.; Scherz, M.D.; Bartonova, A.S.; Pos, D.D. DNA barcodes on their own are not enough to describe a species. Syst. Entomol. 2022, 47, 385–389. [Google Scholar] [CrossRef]
- Will, K.W.; Rubinoff, D. Myth of the molecule: DNA barcodes for species cannot replace morphology for identification and classification. Cladistics 2004, 20, 47–55. [Google Scholar] [CrossRef]
- Hellmuth, M. Biologically feasible gene trees, reconciliation maps and informative triples. Algorithms Mol. Biol. 2017, 12, 23. [Google Scholar] [CrossRef]
- Andujar, C.; Arribas, P.; Yu, D.W.; Vogler, A.P.; Emerson, B.C. Why the COI barcode should be the community DNA metabarcode for the metazoa. Mol. Ecol. 2018, 27, 3968–3975. [Google Scholar] [CrossRef]
- Rubinoff, D.; Cameron, S.; Will, K. Are plant DNA barcodes a search for the Holy Grail? Trends Ecol. Evol. 2006, 21, 1–2. [Google Scholar] [CrossRef]
- Vijayan, K.; Tsou, C.-H. DNA barcoding in plants: Taxonomy in a new perspective. Curr. Sci. 2010, 99, 1530–1541. [Google Scholar]
- Lebonah, D.E.; Dileep, A.; Chandrasekhar, K.; Sreevani, S.; Sreedevi, B.; Pramoda Kumari, J. DNA barcoding on Bacteria: A review. Adv. Biol. 2014, 2014, 541787. [Google Scholar] [CrossRef]
- Xu, J. Fungal DNA barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef]
- Rubinoff, D.; Cameron, S.; Will, K. A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 2006, 97, 581–594. [Google Scholar] [CrossRef]
- Raupach, M.J.; Amann, R.; Wheeler, Q.D.; Roos, C. The application of “-omics” technologies for the classification and identification of animals. Org. Divers. Evol. 2016, 16, 1–12. [Google Scholar] [CrossRef]
- Coissac, E.; Hollingsworth, P.M.; Lavergne, S.; Taberlet, P. From barcodes to genomes: Extending the concept of DNA barcoding. Mol. Ecol. 2016, 25, 1423–1428. [Google Scholar] [CrossRef]
- Guo, B.; Kong, L. Comparing the efficiency of single-locus species delimitation methods within Trochoidea (Gastropoda: Vetigastropoda). Genes 2022, 13, 2273. [Google Scholar] [CrossRef]
- Isaac, N.J.B.; Mallet, J.; Mace, G.M. Taxonomic inflation: Its influence on macroecology and conservation. Trends Ecol. Evol. 2004, 19, 464–469. [Google Scholar] [CrossRef]
- Cotterill, F.P.D.; Taylor, P.J.; Gippoliti, S.; Bishop, J.M.; Groves, C.P. Why one century of phenetics is enough: Response to “Are there really twice as many bovid species as we thought?”. Syst. Biol. 2014, 63, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Primack, R.B. Essentials of Conservation Biology, 6th ed.; Sinauer Associates: Sunderland, MA, USA, 2014; ISBN 978-1605352893. [Google Scholar]
- Zachos, F.E. Taxonomic inflation, the Phylogenetic Species Concept and lineages in the Tree of Life—A cautionary comment on species splitting. J. Zool. Syst. Evol. Res. 2015, 53, 180–184. [Google Scholar] [CrossRef]
- Jaric, I.; Heger, T.; Castro Monzon, F.; Jeschke, J.M.; Kowarik, I.; McConkey, K.R.; Pysek, P.; Sagouis, A.; Essl, F. Crypticity in biological invasions. Trends Ecol. Evol. 2019, 34, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Kotov, A.A.; Karabanov, D.P.; Van Damme, K. Non-indigenous cladocera (Crustacea: Branchiopoda): From a few notorious cases to a potential global faunal mixing in aquatic ecosystems. Water 2022, 14, 2806. [Google Scholar] [CrossRef]
- Vitecek, S.; Kucinic, M.; Previsic, A.; Zivic, I.; Stojanovic, K.; Keresztes, L.; Balint, M.; Hoppeler, F.; Waringer, J.; Graf, W.; et al. Integrative taxonomy by molecular species delimitation: Multi-locus data corroborate a new species of Balkan Drusinae micro-endemics. BMC Evol. Biol. 2017, 17, 129. [Google Scholar] [CrossRef]
- Solovyeva, E.N.; Dunayev, E.A.; Nazarov, R.A.; Bondarenko, D.A.; Poyarkov, N.A. COI-barcoding and species delimitation assessment of toad-headed Agamas of the genus Phrynocephalus (Agamidae, Squamata) reveal unrecognized diversity in Central Eurasia. Diversity 2023, 15, 149. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef]
- Spori, Y.; Stoch, F.; Dellicour, S.; Birky, C.W.; Flot, J.-F. KoT: An automatic implementation of the K/θ method for species delimitation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of Birds through DNA Barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Da Silva, R.; Peloso, P.L.V.; Sturaro, M.J.; Veneza, I.; Sampaio, I.; Schneider, H.; Gomes, G. Comparative analyses of species delimitation methods with molecular data in Snappers (Perciformes: Lutjaninae). Mitochondrial DNA Part A 2018, 29, 1108–1114. [Google Scholar] [CrossRef]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef]
- Jones, G. Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. J. Math. Biol. 2017, 74, 447–467. [Google Scholar] [CrossRef]
- Douglas, J.; Bouckaert, R. Quantitatively defining species boundaries with more efficiency and more biological realism. Commun. Biol. 2022, 5, 755. [Google Scholar] [CrossRef]
- Spori, Y.; Flot, J.-F. HaplowebMaker and CoMa: Two web tools to delimit species using haplowebs and conspecificity matrices. Methods Ecol. Evol. 2020, 11, 1434–1438. [Google Scholar] [CrossRef]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef]
- de Carvalho, M.R.; Bockmann, F.A.; Amorim, D.S.; Brandão, C.R.F.; de Vivo, M.; de Figueiredo, J.L.; Britski, H.A.; de Pinna, M.C.C.; Menezes, N.A.; Marques, F.P.L.; et al. Taxonomic impediment or impediment to taxonomy? A commentary on systematics and the cybertaxonomic-automation paradigm. Evol. Biol. 2007, 34, 140–143. [Google Scholar] [CrossRef]
- Kotov, A.A.; Gololobova, M.A. Traditional taxonomy: Quo vadis? Integr. Zool. 2016, 11, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Dellicour, S.; Flot, J.-F. The hitchhiker’s guide to single-locus species delimitation. Mol. Ecol. Resour. 2018, 18, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Ho, S.Y.W. The molecular clock and evolutionary timescales. Biochem. Soc. Trans. 2018, 46, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Vences, M.; Miralles, A.; Brouillet, S.; Ducasse, J.; Fedosov, A.; Kharchev, V.; Kostdinov, I.; Kumari, S.; Patmanidis, S.; Scherz, M.D.; et al. iTaxoTools 0.1: Kickstarting a specimen-based software toolkit for taxonomists. Megataxa 2021, 6, 77–92. [Google Scholar] [CrossRef]
- Machado, V.N.; Collins, R.A.; Ota, R.P.; Andrade, M.C.; Farias, I.P.; Hrbek, T. One thousand DNA barcodes of piranhas and pacus reveal geographic structure and unrecognised diversity in the Amazon. Sci. Rep. 2018, 8, 8387. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; deWaard, J.R.; Landry, J.-F. DNA barcodes for 1/1000 of the animal kingdom. Biol. Lett. 2010, 6, 359–362. [Google Scholar] [CrossRef]
- Valdez-Moreno, M.; Ivanova, N.V.; Elias-Gutierrez, M.; Contreras-Balderas, S.; Hebert, P.D.N. Probing diversity in freshwater fishes from Mexico and Guatemala with DNA barcodes. J. Fish Biol. 2009, 74, 377–402. [Google Scholar] [CrossRef]
- Pereira, L.H.G.; Hanner, R.; Foresti, F.; Oliveira, C. Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? BMC Genet. 2013, 14, 20. [Google Scholar] [CrossRef]
- Turanov, S.V.; Kartavtsev, Y.P. A complement to DNA barcoding reference library for identification of fish from the Northeast Pacific. Genome 2021, 64, 927–936. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Bekker, E.I.; Pavlov, D.D.; Borovikova, E.A.; Kodukhova, Y.V.; Kotov, A.A. New sets of primers for DNA identification of non-indigenous fish species in the Volga-Kama basin (European Russia). Water 2022, 14, e437. [Google Scholar] [CrossRef]
- Pavlov, D.S. (Ed.) Red Book of the Russian Federation, 2nd ed.; Volume “Animals”; FGBU “VNII Ecologiya”: Moscow, Russia, 2021; ISBN 978-5-6047425-0-1. [Google Scholar]
- Kodukhova, Y.V.; Karabanov, D.P. Morphological changes in the population of roach (Rutilus rutilus, Cyprinidae) in lake Pleshcheevo as a result of the introduction of the mollusk, Dreissena polymorpha (Bivalvia). Zool. Zhurnal 2017, 96, 1069–1077. [Google Scholar] [CrossRef]
- Smirnov, A.K.; Pavlov, D.D.; Kodukhova, Y.V.; Karabanov, D.P. Impact of Zebra mussel Dreissena polymorpha pallas 1771 (Bivalvia) appearance on fish populations in Lake Pleshcheevo, European Russia. Zool. Zhurnal 2020, 99, 1363–1374. [Google Scholar] [CrossRef]
- Rosstat. Population Census: Official statistics. Available online: https://eng.rosstat.gov.ru/folder/76215 (accessed on 24 February 2023).
- Pravdin, I.F. Guide for the Study of Fish; Pishchevaya Promyshlennost: Moscow, Russia, 1966. [Google Scholar]
- Kottelat, M.; Freyhof, J. Handbook of European Freshwater Fishes; Publications Kottelat: Cornol, Switzerland, 2007; ISBN 2839902982. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Birstein, V.J.; DeSalle, R.; Doukakis, P.; Hanner, R.; Ruban, G.I.; Wong, E. Testing taxonomic boundaries and the limit of DNA barcoding in the Siberian sturgeon, Acipenser baerii. Mitochondrial DNA 2009, 20, 110–118. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schaffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Froese, R.; Pauly, D. (Eds.) FishBase. World Wide Web Electronic Publication, Version 02/2023. Available online: www.fishbase.org (accessed on 23 February 2023).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Le Vinh, S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; ISBN 0195135857. [Google Scholar]
- Goloboff, P.A.; Catalano, S.A. TNT version 1.5, including a full implementation of phylogenetic morphometrics. Cladistics 2016, 32, 221–238. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; de Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Barido-Sottani, J.; Boskova, V.; Du Plessis, L.; Kuhnert, D.; Magnus, C.; Mitov, V.; Muller, N.F.; PecErska, J.; Rasmussen, D.A.; Zhang, C.; et al. Taming the BEAST—A community teaching material resource for BEAST2. Syst. Biol. 2018, 67, 170–174. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST2; Cambridge University Press: Cambridge, UK, 2015; ISBN 978-1-107-01965-2. [Google Scholar]
- Floyd, R.; Abebe, E.; Papert, A.; Blaxter, M. Molecular barcodes for soil nematode identification. Mol. Ecol. 2002, 11, 839–850. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Yang, C.; Zheng, Y.; Tan, S.; Meng, G.; Rao, W.; Yang, C.; Bourne, D.G.; O’Brien, P.A.; Xu, J.; Liao, S.; et al. Efficient COI barcoding using high throughput single-end 400 bp sequencing. BMC Genom. 2020, 21, 862. [Google Scholar] [CrossRef]
- Birky, C.W. Species detection and identification in sexual organisms using population genetic theory and DNA sequences. PLoS ONE 2013, 8, e52544. [Google Scholar] [CrossRef]
- Rangel-Medrano, J.D.; Ortega-Lara, A.; Marquez, E.J. Ancient genetic divergence in bumblebee catfish of the genus Pseudopimelodus (Pseudopimelodidae: Siluriformes) from northwestern South America. PeerJ 2020, 8, e9028. [Google Scholar] [CrossRef]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.-C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Collins, R.A.; Boykin, L.M.; Cruickshank, R.H.; Armstrong, K.F. Barcoding’s next top model: An evaluation of nucleotide substitution models for specimen identification. Methods Ecol. Evol. 2012, 3, 457–465. [Google Scholar] [CrossRef]
- Ward, R.D. DNA barcode divergence among species and genera of birds and fishes. Mol. Ecol. Resour. 2009, 9, 1077–1085. [Google Scholar] [CrossRef]
- Kartavtsev, Y.P. Sequence divergence at mitochondrial genes in animals: Applicability of DNA data in genetics of speciation and molecular phylogenetics. Mar. Genom. 2011, 4, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ota, R.P.; Machado, V.N.; Andrade, M.C.; Collins, R.A.; Farias, I.P.; Hrbek, T. Integrative taxonomy reveals a new species of pacu (Characiformes: Serrasalmidae: Myloplus) from the Brazilian Amazon. Neotrop. Ichthyol. 2020, 18, e190112. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Reid, N.M.; Carstens, B.C. Phylogenetic estimation error can decrease the accuracy of species delimitation: A Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol. Biol. 2012, 12, 196. [Google Scholar] [CrossRef]
- Kotov, A.A.; Garibian, P.G.; Bekker, E.I.; Taylor, D.J.; Karabanov, D.P. A new species group from the Daphnia curvirostris species complex (Cladocera: Anomopoda) from the eastern Palaearctic: Taxonomy, phylogeny and phylogeography. Zool. J. Linn. Soc. 2021, 191, 772–822. [Google Scholar] [CrossRef]
- Talavera, G.; Dinca, V.; Vila, R. Factors affecting species delimitations with the GMYC model: Insights from a butterfly survey. Methods Ecol. Evol. 2013, 4, 1101–1110. [Google Scholar] [CrossRef]
- Lohse, K. Can mtDNA barcodes be used to delimit species? A response to Pons et al. (2006). Syst. Biol. 2009, 58, 439–442; Discussion 442–444. [Google Scholar] [CrossRef]
- Neretina, A.N.; Karabanov, D.P.; Sacherova, V.; Kotov, A.A. Unexpected mitochondrial lineage diversity within the genus Alonella Sars, 1862 (Crustacea: Cladocera) across the Northern Hemisphere. PeerJ 2021, 9, e10804. [Google Scholar] [CrossRef]
- Xu, R.; Lu, Y.; Tang, Y.; Chen, Z.; Xu, C.; Zhang, X.; Zheng, X. DNA barcoding reveals high hidden species diversity of Chinese waters in the Cephalopoda. Front. Mar. Sci. 2022, 9, 830381. [Google Scholar] [CrossRef]
- Sukumaran, J.; Knowles, L.L. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 2017, 114, 1607–1612. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Kodukhova, Y.V.; Pashkov, A.N.; Reshetnikov, A.N.; Makhrov, A.A. “Journey to the West”: Three phylogenetic lineages contributed to the invasion of Stone Moroko, Pseudorasbora parva (Actinopterygii: Cyprinidae). Russ. J. Biol. Invasions 2021, 12, 67–78. [Google Scholar] [CrossRef]
- Gu, Q.; Wang, S.; Zhong, H.; Yuan, H.; Yang, J.; Yang, C.; Huang, X.; Xu, X.; Wang, Y.; Wei, Z.; et al. Phylogeographic relationships and the evolutionary history of the Carassius auratus complex with a newly born homodiploid raw fish (2nNCRC). BMC Genom. 2022, 23, 242. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Bekker, E.I.; Kotov, A.A. Underestimated consequences of biological invasions in phylogeographic reconstructions as seen in Daphnia magna (Crustacea, Cladocera). Zool. Zhurnal 2020, 99, 1232–1241. [Google Scholar] [CrossRef]
- Ducasse, J.; Ung, V.; Lecointre, G.; Miralles, A. LIMES: A tool for comparing species partition. Bioinformatics 2020, 36, 2282–2283. [Google Scholar] [CrossRef]
- Miralles, A.; Vences, M. New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in Madascincus lizards. PLoS ONE 2013, 8, e68242. [Google Scholar] [CrossRef]
- Ahrens, D.; Fujisawa, T.; Krammer, H.-J.; Eberle, J.; Fabrizi, S.; Vogler, A.P. Rarity and incomplete sampling in DNA-based species delimitation. Syst. Biol. 2016, 65, 478–494. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Heatmap3: An improved heatmap package with more powerful and convenient features. BMC Bioinform. 2014, 15, P16. [Google Scholar] [CrossRef]
- Box, G.E.P. Science and Statistics. J. Am. Stat. Assoc. 1976, 71, 791–799. [Google Scholar] [CrossRef]
- Steinegger, M.; Salzberg, S.L. Terminating contamination: Large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 2020, 21, 115. [Google Scholar] [CrossRef]
- Pentinsaari, M.; Ratnasingham, S.; Miller, S.E.; Hebert, P.D.N. BOLD and GenBank revisited—Do identification errors arise in the lab or in the sequence libraries? PLoS ONE 2020, 15, e0231814. [Google Scholar] [CrossRef] [PubMed]
- Stolbunov, I.A.; Dien, T.D.; Karabanov, D.P. Taxonomic composition and distribution of alien suckermouth armored Catfish (Siluriformes: Loricariidae) in Southern Vietnam. Inland Water Biol. 2021, 14, 263–273. [Google Scholar] [CrossRef]
- Ward, R.D.; Hanner, R.; Hebert, P.D.N. The campaign to DNA barcode all fishes, FISH-BOL. J. Fish Biol. 2009, 74, 329–356. [Google Scholar] [CrossRef] [PubMed]
- Lajbner, Z.; Linhart, O.; Kotlik, P. Human-aided dispersal has altered but not erased the phylogeography of the tench. Evol. Appl. 2011, 4, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, Y.V.; Smirnov, A.K.; Kodukhova, Y.V. Assessment of possible causes of changes in abundance and sexual structure in populations of Prussian Carp (Carassius auratus gibelio Bloch, 1783). Inland Water Biol. 2018, 11, 72–80. [Google Scholar] [CrossRef]
- Denys, G.P.J.; Dettai, A.; Persat, H.; Hautecœur, M.; Keith, P. Morphological and molecular evidence of three species of pikes Esox spp. (Actinopterygii, Esocidae) in France, including the description of a new species. C. R. Biol. 2014, 337, 521–534. [Google Scholar] [CrossRef]
- Dyldin, Y.V.; Hanel, L.; Fricke, R.; Orlov, A.M.; Romanov, V.I.; Plesnik, J.; Interesova, E.A.; Vorobiev, D.S.; Kochetkova, M.O. Fish diversity in freshwater and brackish water ecosystems of Russia and adjacent waters. Publ. Seto Mar. Biol. Lab. 2020, 45, 47–116. [Google Scholar] [CrossRef]
- Borovikova, E.; Nikulina, Y. The contact zone of phylogenetic lineages of freshwater fish in Arctic Eurasia: Genetic polymorphism of Coregonid populations. Diversity 2023, 15, 163. [Google Scholar] [CrossRef]
- Svetovidov, A.N. Salmonidae. In Fishes of the North-Eastern Atlantic and the Mediterranean; Whitehead, P.J., Bauchot, M.L., Hureau, J.C., Nielsen, J., Tortonese, E., Eds.; UNESCO: Paris, France, 1984; Volume 1, pp. 373–385. ISBN 9230022152. [Google Scholar]
- Borovikova, E.A.; Artamonova, V.S. Vendace (Coregonus albula) and least cisco (Coregonus sardinella) are a single species: Evidence from revised data on mitochondrial and nuclear DNA polymorphism. Hydrobiologia 2021, 848, 4241–4262. [Google Scholar] [CrossRef]
- Kodukhova, Y.V.; Karabanov, D.P. Finding of Longtail Dwarf Goby Knipowitschia longecaudata (Actinopterygii: Gobiidae) in the upper part of unregulated section of the Volga River. Inland Water Biol. 2021, 14, 620–625. [Google Scholar] [CrossRef]
- Zachos, F.E. Species Concepts in Biology. Historical Development, Theoretical Foundations and Practical Relevance; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-44964-7. [Google Scholar]
- Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. B 2005, 360, 1935–1943. [Google Scholar] [CrossRef]
- Avise, J.C.; Walker, D. Species realities and numbers in sexual vertebrates: Perspectives from an asexually transmitted genome. Proc. Natl. Acad. Sci. USA 1999, 96, 992–995. [Google Scholar] [CrossRef]
- Minelli, A. Taxonomy needs pluralism, but a controlled and manageable one. Megataxa 2020, 1, 9–18. [Google Scholar] [CrossRef]
- Mina, M.V.; Reshetnikov, Y.S.; Dgebuadze, Y.Y. Taxonomic novelties and problems for users. J. Ichthyol. 2006, 46, 476–480. [Google Scholar] [CrossRef]
- Mina, M.V. Should ichthyologists abandon the concept of “polymorphic species”? In Actual Problems of Modern Ichthyology (to the 100th Anniversary of G.V. Nikolsky); Pavlov, D.S., Dgebuadze, Y.Y., Shatunovskii, M.I., Eds.; KMK Scientific Press Ltd.: Moscow, Russia, 2010; pp. 88–95. ISBN 978-5-87317-643-4. (In Russian) [Google Scholar]
- Nikolsky, G.V. Structure of the Species and Patterns of Fish Variability; Food Industry: Moscow, Russia, 1980. [Google Scholar]
- Mayr, E.; Ashlock, P.D. Principles of Systematic Zoology, 2nd ed.; McGraw-Hill: New York, NY, USA, 1991; ISBN 0070411441. [Google Scholar]
- Kunz, W. Diversity within the species: Polymorphisms and the polytypic species. In Do Species Exist? Principles of Taxonomic Classification; Kunz, W., Ed.; Wiley-Blackwell: Weinheim, Germany, 2012; pp. 93–126. ISBN 9783527332076. [Google Scholar]
- Ross, H.H.; Decker, G.C.; Cunningham, H.B. Adaptation and differentiation of temperate phylogenetic lines from tropical ancestors in Empoasca. Evolution 1964, 18, 639–651. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Kodukhova, Y.V. Biochemical polymorphism and intraspecific structure in populations of Kilka Clupeonella cultriventris (Nordmann, 1840) from natural and invasive parts of its range. Inland Water Biol. 2018, 11, 496–500. [Google Scholar] [CrossRef]
- Jaenike, J. Criteria for ascertaining the existence of host races. Am. Nat. 1981, 117, 830–834. [Google Scholar] [CrossRef]
- ICZN. International Code of Zoological Nomenclature, 4th ed.; International Trust for Zoological Nomenclature: London, UK, 1999; ISBN 0853010064. [Google Scholar]
- Borovikova, E.A. Special traits of the genetic structure and origin of the population of vendace Coregonus albula of Pleshcheyevo Lake. Biol. Bull. 2017, 44, 245–250. [Google Scholar] [CrossRef]
- Mina, M.V. Problems of protection of fish faunas in the USSR. Neth. J. Zool. 1991, 42, 200–213. [Google Scholar] [CrossRef]
- Funk, D.J.; Omland, K.E. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with Insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 397–423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| GB | BN | KoT | SI | LM | AB | AS | YC | bYC | PT | bPT | mPT | STC | SPD | HwM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GB | 60 | 68 | 69 | 62 | 67 | 67 | 67 | 75 | 67 | 64 | 68 | 71 | 79 | 47 | |
| BN | 52 | 47 | 46 | 42 | 47 | 45 | 41 | 47 | 47 | 44 | 47 | 48 | 55 | 42 | |
| KoT | 50 | 35 | 72 | 83 | 86 | 79 | 69 | 80 | 86 | 60 | 89 | 84 | 85 | 40 | |
| SI | 55 | 37 | 62 | 60 | 77 | 4 | 76 | 72 | 84 | 77 | 72 | 68 | 76 | 52 | |
| LM | 44 | 29 | 74 | 43 | 71 | 94 | 58 | 83 | 71 | 50 | 83 | 88 | 79 | 36 | |
| AB | 51 | 36 | 83 | 67 | 58 | 76 | 81 | 77 | 91 | 70 | 86 | 81 | 82 | 47 | |
| AS | 52 | 36 | 67 | 47 | 91 | 62 | 62 | 89 | 76 | 53 | 89 | 94 | 84 | 38 | |
| YC | 58 | 33 | 61 | 72 | 42 | 73 | 45 | 69 | 81 | 87 | 69 | 65 | 67 | 55 | |
| bYC | 68 | 42 | 68 | 58 | 74 | 63 | 83 | 61 | 77 | 60 | 80 | 94 | 85 | 43 | |
| PT | 51 | 36 | 88 | 75 | 63 | 86 | 67 | 73 | 68 | 70 | 86 | 81 | 82 | 47 | |
| bPT | 50 | 25 | 48 | 70 | 34 | 57 | 38 | 79 | 48 | 60 | 60 | 57 | 63 | 64 | |
| mPT | 50 | 35 | 84 | 62 | 74 | 78 | 83 | 57 | 68 | 83 | 48 | 84 | 85 | 40 | |
| STC | 60 | 43 | 76 | 50 | 82 | 70 | 91 | 53 | 92 | 75 | 41 | 76 | 89 | 40 | |
| SPD | 67 | 53 | 77 | 65 | 67 | 71 | 76 | 55 | 77 | 76 | 43 | 77 | 84 | 45 | |
| HwM | 26 | 18 | 16 | 35 | 14 | 18 | 17 | 29 | 19 | 25 | 46 | 19 | 16 | 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karabanov, D.P.; Kotov, A.A.; Borovikova, E.A.; Kodukhova, Y.V.; Zhang, X. Comparison of the Efficiency of Single-Locus Species Delimitation Methods: A Case Study of a Single Lake Fish Population in Comparison against the Barcodes from International Databases. Water 2023, 15, 1851. https://doi.org/10.3390/w15101851
Karabanov DP, Kotov AA, Borovikova EA, Kodukhova YV, Zhang X. Comparison of the Efficiency of Single-Locus Species Delimitation Methods: A Case Study of a Single Lake Fish Population in Comparison against the Barcodes from International Databases. Water. 2023; 15(10):1851. https://doi.org/10.3390/w15101851
Chicago/Turabian StyleKarabanov, Dmitry P., Alexey A. Kotov, Elena A. Borovikova, Yulia V. Kodukhova, and Xiaowei Zhang. 2023. "Comparison of the Efficiency of Single-Locus Species Delimitation Methods: A Case Study of a Single Lake Fish Population in Comparison against the Barcodes from International Databases" Water 15, no. 10: 1851. https://doi.org/10.3390/w15101851