
Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis

1
School of Life Science, Hefei Normal University, Hefei 230601, China
2
National Positioning Observation and Research Station of the Qinghai Lake Wetland Ecosystem in Qinghai Province, National Forestry and Grassland Administration, Haibei Tibetan Autonomous Prefecture 812305, China
3
College of The Ecology and Environment, Xinjiang University, Urumqi 830046, China
*
Author to whom correspondence should be addressed.
Water 2023, 15(2), 239; https://doi.org/10.3390/w15020239
Submission received: 16 November 2022
/
Revised: 30 December 2022
/
Accepted: 1 January 2023
/
Published: 5 January 2023
(This article belongs to the Special Issue Advances in Forest Hydrology and Biogeochemistry)
Abstract
:Poa alpigena Lindm. is a dominant forage grass that is widely distributed on the Qinghai-Tibetan Plateau and is often used in the restoration of degraded grasslands. Soil microorganisms are major players in the cycling of materials in terrestrial ecosystems. In this study, based on high-throughput sequencing, the rhizosphere and non-rhizosphere soils of Poa alpigena L. on Bird Island, Qinghai Lake, were used to investigate the effects of Poa alpigena L. on the composition and structure of soil microbial communities, and to establish associated soil microbial gene pools. Results revealed that microorganisms in the soil of Poa alpigena L. on Bird Island belonged to 62 phyla, 112 classes, 245 orders, 518 families, 1610 genera, and 5704 species. The dominant soil bacteria in rhizosphere and non-rhizosphere soils were Proteobacteria (49.62%, 47.13%) and Actinobacteria (30.31% and 31.67%), whereas the dominant fungi were Ascomycota (3.15% and 3.37%) and Basidiomycota (0.98% and 1.06%). Alpha diversity analysis revealed that the microbial richness and diversity in non-rhizosphere soil were significantly higher than those in rhizosphere soil, mainly influenced by soil water content and total nitrogen content. Furthermore, on the basis of LEfSe analysis, Alphaproteobacteria and Betaproteobacteria were identified as prominent differential taxa for rhizosphere and non-rhizosphere soils, respectively. The key differential metabolic pathways of rhizosphere soil microorganisms were those associated with the ATP-binding cassette (ABC) transporter, basal metabolism, and cytochrome P450 metabolism, whereas those of non-rhizosphere soil microorganisms included the gene expression-related pathways, methane metabolism, and pathway associated with degradation of aromatic compounds. These findings indicated that the rhizosphere soil of Poa alpigena L. is selective for microorganisms that play important roles in the oxidation of methane and regulation of the greenhouse effect on Bird Island, and that the soil environment on this island may be subject to contamination with aromatic compounds.
1. Introduction
As important components of soil ecosystems, soil microorganisms play key roles in ecosystem material cycling, affecting a diverse range of ecosystem processes, including soil carbon and nitrogen cycling, plant productivity, and litter decomposition, and the microbial species and functional diversity can serve as important indicators for evaluating soil quality [1,2,3]. The rhizosphere is one of the sites of particularly heightened plant material cycling activity and energy metabolism, and rhizosphere microorganisms play key roles in plant growth and development, which can improve plant productivity and resistance as well as soil properties, ultimately leading to changes in plant communities and soil ecosystem elemental cycles and patterns [4,5]. Rhizosphere microorganisms have positive effects on plant growth, soil fertility, and the overall health of the soil’s ecological environment [6].
The Qinghai Lake basin, which is a typical fragile ecosystem and an area sensitive to global change, represents a natural barrier that contributes to maintaining the ecological security of the northeastern part of the Tibetan Plateau and prevents the spread of western desertification to the east, and is acknowledged to be an area rich in gene diversity and biodiversity [7]. Alternating land-use patterns and grazing intensity in the Qinghai Lake basin have marked effects on soil nutrient content, resulting in varying degrees of soil degradation and, consequently, changes in the metabolic characteristics of the soil microbial community [8,9]. In recent years, studies on the population structure of rhizosphere soil microorganisms and their associated changes have accordingly become a particular focus of ecological research in this area.
Poa alpigena Lindm., with characteristics of cold and drought resistance, is an important dominant forage plant in the alpine meadows of the Qinghai Lake region, and has been widely used in livestock production and the restoration of degraded grassland in this region [10]. At present, research on Poa alpigena L. and Poa pratensis have tended to focus on individual traits, community diversity, adaptability, productivity, stress tolerance, and soil nutrients, mainly with respect to Poa pratensis [10,11,12,13]. However, there have been comparatively few studies that have sought to examine the soil microorganisms associated with P. alpigena in the Qinghai Lake basin. Nevertheless, studying the rhizosphere soil microbial community of Poa alpigena L. is of particular importance from the perspectives of alpine grassland conservation, desert management, and ecological restoration. Consequently, in this study, metagenomics combined with bioinformatics was used to analyze the differences in the structure, diversity, and metabolic pathways of rhizosphere and non-rhizosphere soil microbial communities of Poa alpigena L. on Bird Island in Qinghai Lake. The aim of this study was to establish a database of soil microbial resources for the grassland ecosystem of the Bird Island Nature Reserve, to provide a scientific basis for accelerating the restoration of ecological functions of degraded grassland, and promote the sustainable development of the Qinghai Lake area.
2. Materials and Methods
2.1. Study Area
Bird Island is located in the northwest sector of Qinghai Lake (36°57′–37°04′ N, 99°44′–99°54′ E). It comprises an area of lakeside wetland with an elevation of 3194–3226 m and has been established as the Bird Island National Nature Reserve. The terrain is high in the northwest and low in the southeast. The average annual temperature of the region is −0.7 °C, with maximum and minimum temperatures of 28 °C and −31 °C, respectively, and the average annual precipitation is 322.7 mm. The area lies within the semi-arid and alpine climatic zone of the plateau and the surface vegetation comprises predominantly Poa alpigena L., Stipa purpurea Griseb., Carex rigescens, Leymus secalinus, Polygonum sibiricum Laxm., Allium przewalskianum, and Astragalus adsurgens Pall.
2.2. Sample Treatment
The fenced and undisturbed Poa alpigena L. community in the National Positioning Observation and Research Station of the Qinghai Lake Wetland Ecosystem (Bird Island Station) was selected as the research object on Bird Island. Four parallel plots of 2 m × 2 m were randomly set in this community, and each plot was more than 20 m away from the nearest neighboring plot. Within each of these plots, five Poa alpigena L. plants of the same developmental status were selected according to an “S” type route. The rhizosphere soil of each plant was collected using the shake-off method, described by Riley and Barber [14,15], from each plot. The weakly adhering soil was gently shaken off and discarded. The soil remaining strongly adhered to roots (0–1 mm) was brushed off with a sterile soft brush and subsequently recorded as rhizosphere soil (NG). The non-rhizosphere soil was obtained from the 0–20 cm vertical surface within the projection range of plant rhizosphere (NC). Four rhizosphere and four non-rhizosphere soil samples were then individually placed in sterile bottles and marked as follows: NG1, NG2, NG3, and NG4 for the rhizosphere soil samples, and NC1, NC2, NC3, and NC4 for the non-rhizosphere soil samples. All samples were divided into two parts after removing roots, leaves, and stones. One part was immediately stored in liquid nitrogen, returned to the laboratory, and stored at −80 °C for DNA extraction and meta-genomics sequencing. The other part was brought back to the laboratory and used to measure water content, total nitrogen, total carbon, pH, and conductivity. During the sampling process, the samplers wore sterile masks and sterile gloves, excavated the whole plant with a sterile soil scooper (13 cm × 40 cm), and brushed the rhizosphere soil with a sterile soft brush. Having collected each sample, masks, gloves, and brushes were replaced, and the scooper was disinfected with 75% alcohol.
Soil water content was determined using a soil moisture meter (JK-100F) with an accuracy of 0.1%, total carbon (TC) was determined using a CE-440 elemental analyzer (EAI., Chicago, IL, USA), and total nitrogen (TN) was determined using the Kjeldahl method. Soil pH (1:2.5 soil/water suspension) and conductivity (1:5 for soil/water leaching solution) were determined using pHS-25 (precision of 0.05) and DDS-307 (precision of 0.01), respectively.
Soil DNA extraction and purification methods were referenced from Han et al. [16], using chloroform-isoamyl alcohol to extract crude DNA, isopropanol to re-precipitate DNA, and a QIAquick Gel Extraction Kit buffer to purify the DNA. The concentration of DNA in the samples was measured using a Qubit Fluorometer and the integrity of the DNA was determined using 1% agarose gel electrophoresis. After detecting the concentration and integrity of the DNA, DNA samples were sent to BGI to perform metagenomics high-throughput sequencing using the BGISEQ-500 sequencing platform. All DNAs in samples, including bacterial, archaeal, fungal, viral, and protozoan DNA, were interrupted into cDNA fragments (∼300 bp). Paired-end sequencing of cDNA fragments (∼300 bp) was conducted using the BGISEQ-500 sequencing platform by BGI-Shenzhen, China, to obtain the raw data.
2.3. Data Analysis
To obtain high-quality valid sequences, the raw sequencing data were processed using Trimmomatic software (v3.3) to remove linker sequences, etc., after which the experiments were performed using the original default parameters. The cleaned linker sequences were as follows:
PrefixPE/1: AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA;
PrefixPE/2: AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG.
Metagenomics assembly was performed using Megahit software with default parameters and combining all sample sequences. The results of the metagenomics assembly were evaluated using Metaquast software to compare the assembly results with the reference sequence and to obtain information on the number of high-quality contigs, the longest contig, and N50 of the assembled sequences.
Sequences were compared and analyzed with constructed species composition databases (downloaded from NCBI databases including bacterial, archaebacterial, fungal, viral, protozoan genomes, and NT databases) using Kraken v2 software. The classification report of DNA sequences was performed by importing the Kraken data file to the supporting Pavian R package v1.2.0. Species annotations and determinations of relative abundances were performed using Bracken software in combination with taxonomic information obtained from NCBI, with data being classified at the phylum, class, order, family, genus, and species levels. Alpha diversity analysis was performed using Vegan software in the R package and beta diversity was analyzed based on principal coordinates analysis (PCoA) using R software. Indicator microorganisms and metabolic pathways were identified by a least discriminant analysis (LDA) effect size (LEfSe) analysis of raw intergroup microbial data and cumulative metabolic pathway data (KEGG analysis), respectively.
Statistical analysis of the data was performed using SPSS 21.0. Differences between rhizosphere and non-rhizosphere soil with respect to soil properties, microbial sequences, and α-diversity were detected by a t-test. Simple linear regression analysis with a t-test was used to assess the relationships between soil properties and α-diversity indexes.
The metagenomics sequencing data acquired in this study are available at the National Center for Biotechnology Information, USA, (https://www.ncbi.nlm.nih.gov/sra/PRJNA867494).
3. Results
3.1. Physicochemical Properties of Rhizosphere and Non-Rhizosphere Soils
The physicochemical properties of rhizosphere and non-rhizosphere soil of Poa alpigena L. on Bird Island were shown in Table 1. The water content and total nitrogen content of rhizosphere soil were significantly lower than those of non-rhizosphere soil (p < 0.05). Similarly, the pH of rhizosphere soil was lower than that of non-rhizosphere, but not significantly, and the differences in other physicochemical properties were small.
3.2. Microbial Composition of Poa alpigena L. Soils on Bird Island
The metagenomics sequencing results showed that for each sample, in excess of 10 GB of sequence data, the average number of sequences reaching more than 36 million pairs were obtained. These data indicated that a sufficient level of sequencing for microbiota in the soil samples of Poa alpigena L. was achieved and that the results showed little fluctuation. Moreover, approximately 54–58% of the detected sequences were not classified as existing sequences of known species (Table 2). The percentage of microbial sequences in non-rhizosphere soils was 35.97–37.47%, with bacteria accounting for 28.80%. The percentage of microbial sequences in rhizosphere soils was 36.90–41.07%, which was significantly higher than that in non-rhizosphere soils, with bacteria reaching 32.22%, slightly higher than that of non-rhizosphere soils. Fungal, viral, and protozoan accounted for 1.42%, 0.15%, and 0.17% of the total sequences in both rhizosphere and non-rhizosphere soils, respectively, with no significant difference.
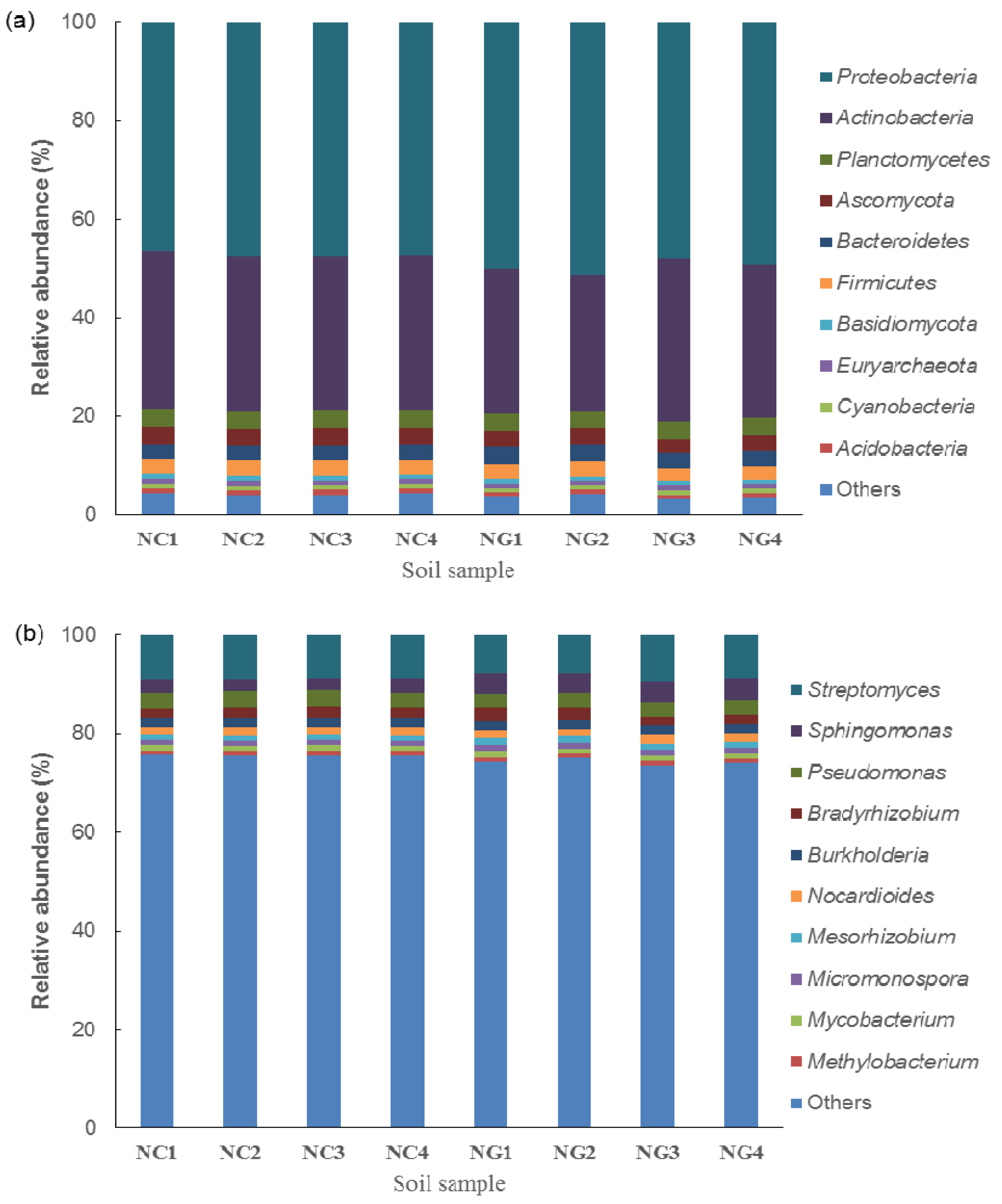
The soil microbial composition showed high diversity in all soil samples. The microorganisms of the rhizosphere and non-rhizosphere soil samples belonged to 62 phyla, 112 classes, 245 orders, 518 families, 1610 genera, and 5704 species, the relative abundance of which were shown in Figure 1 and Figure S1 (Supplementary material).
At the phylum level, as shown in Figure 1a, Proteobacteria was the dominant phylum with the highest average relative abundance of 49.62% and 47.13% in rhizosphere and non-rhizosphere soils, respectively, followed by Actinobacteria, with an average relative abundance of 30.31% and 31.67%. Among the fungi, Ascomycota and Basidiomycota were the dominant phyla in rhizosphere and non-rhizosphere soils. The average relative abundance of Ascomycota was 3.15% and 3.37% in rhizosphere and non-rhizosphere soils, respectively. The average relative abundance of Basidiomycota was 0.98% and 1.06% in rhizosphere and non-rhizosphere soils.
At the genus level (Figure 1b), Streptomyces had the highest average relative abundance of 8.43% and 8.90% in rhizosphere and non-rhizosphere soils, respectively. This was followed by Sphingomonas, with an average relative abundance of 4.13% and 2.59%, and Pseudomonas, with an average relative abundance of 3.03% and 3.30%, respectively.
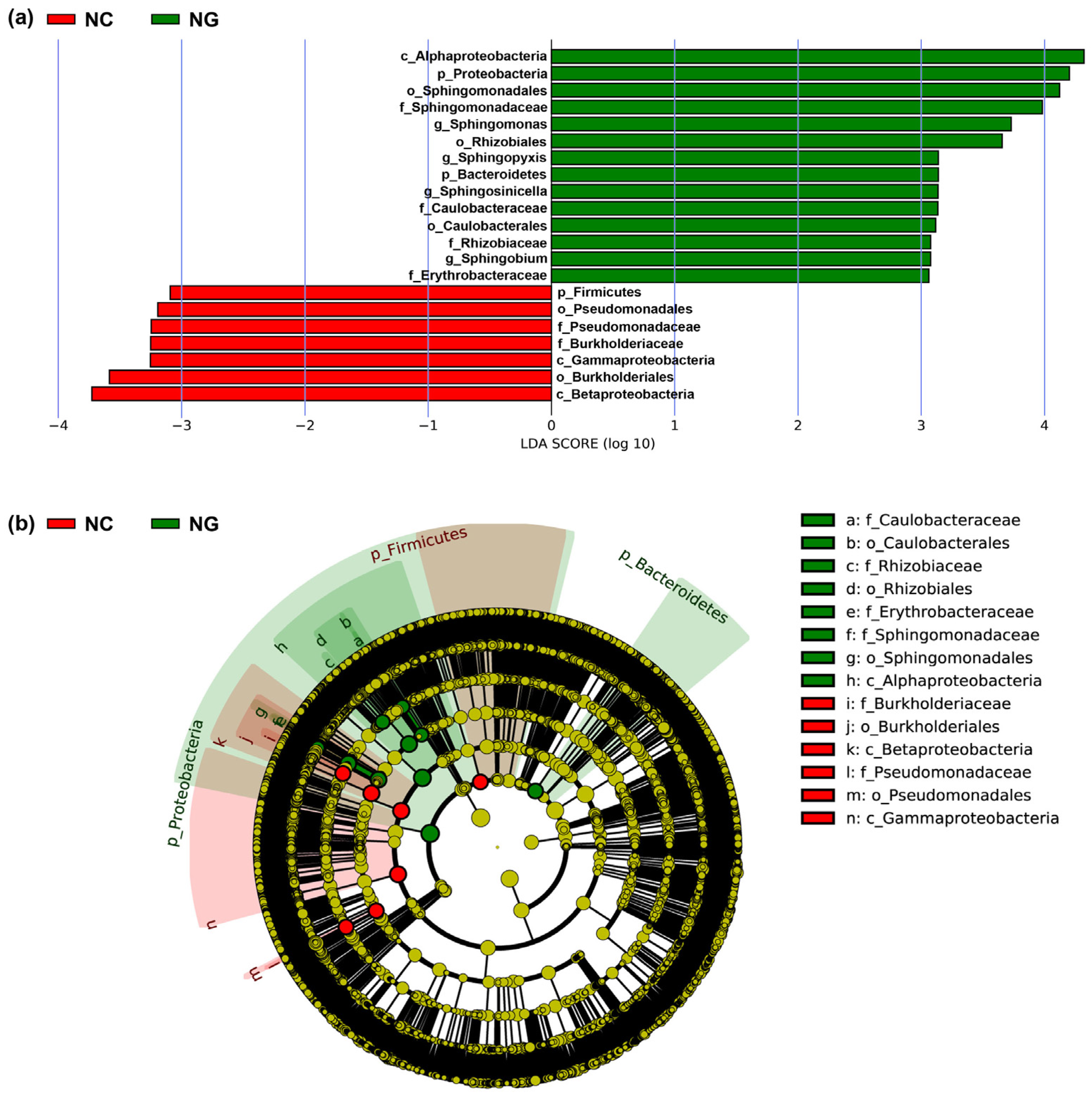
To further analyze microbial community structure in rhizosphere and non-rhizosphere soils, LEfSe analysis was performed, which showed only those differential microbes that met the linear discriminant analysis (LDA) significance threshold of >3.0 (Figure 2). Accordingly, a total of 21 differential microbes in rhizosphere (14) and non-rhizosphere soils (7) were identified. The differential microbes in rhizosphere soils mainly included Proteobacteria, Alphaproteobacteria, Sphingomonadales, Rhizobiales, Sphingomonadaceae, and Sphingomonas. The differential microbes in non-rhizosphere soils mainly included Betaproteobacteria, Gammaproteobacteria, Burkholderiales, Pseudomonadales, Burkholderiaceae, and Pseudomonadaceae. Among these, the largest contributors in rhizosphere and non-rhizosphere soils were Alphaproteobacteria and Betaproteobacteria, respectively, which could accordingly be used as indicator microorganisms for the two soil types.
3.3. Microbial Diversity of Poa alpigena L. Soils on Bird Island
3.3.1. Microbial α-Diversity
Analysis of microbial α-diversity revealed notable differences between the rhizosphere and non-rhizosphere soils of Poa alpigena L. on Bird Island. As shown in Table 3, values of the Richness index, Chao 1 index, and Shannon index (5702.00, 5703.18, and 7.78) of non-rhizosphere soil microorganisms were significantly higher than those of rhizosphere soil microorganisms (5654.75, 5655.98, and 7.73). The Simpson index value in non-rhizosphere soil (1.10 × 10−3) was significantly lower than that in rhizosphere soil microorganisms (1.15 × 10−3). These results indicated that the richness and diversity of non-rhizosphere soil microorganisms were significantly higher than those of rhizosphere soil microorganisms.
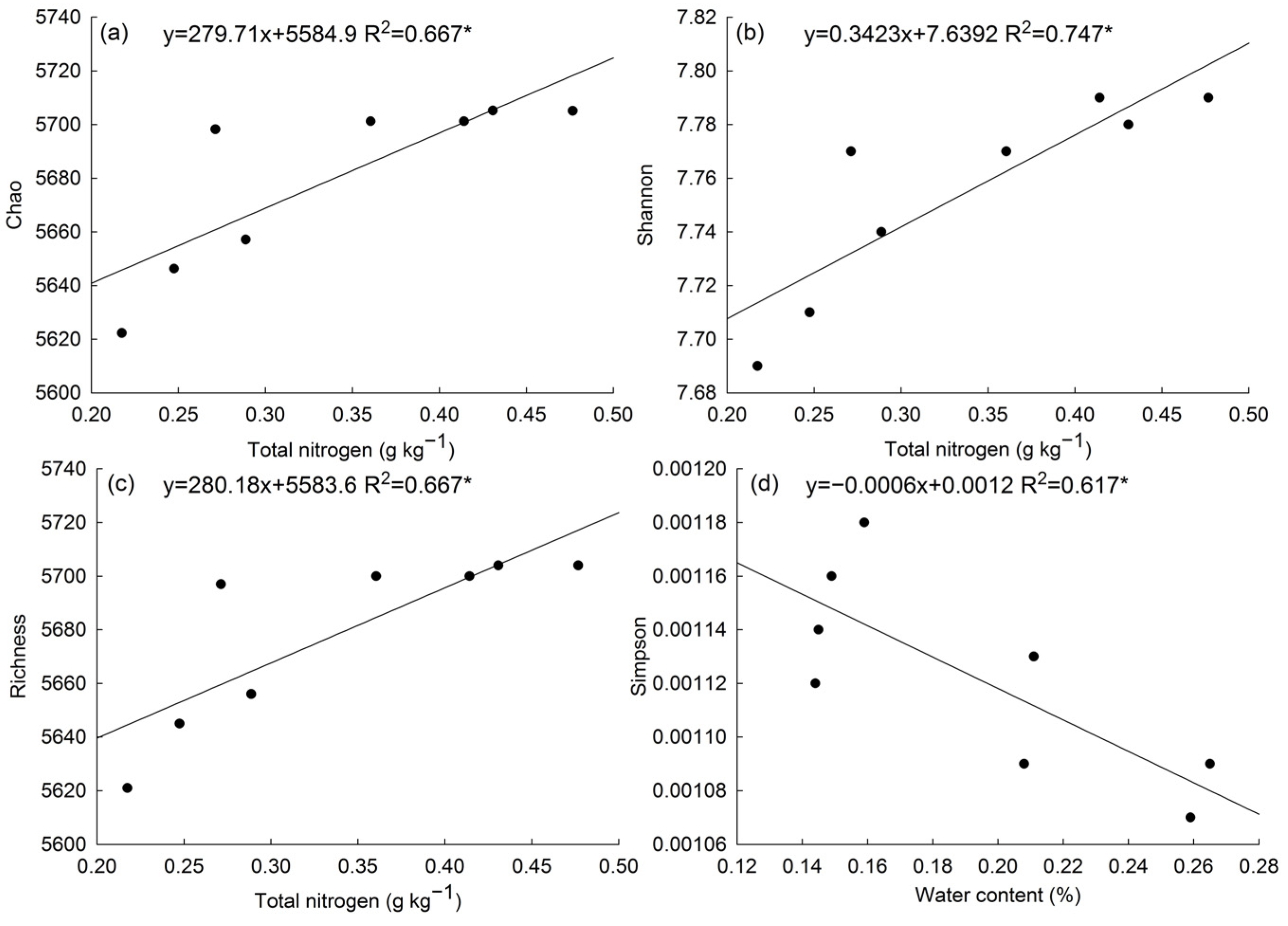
To further investigate whether soil microbial diversity was associated with soil physicochemical properties, a regression analysis was performed, for which soil physicochemical properties and microbial community diversity indexes were used as explanatory and response variables, respectively (Figure 3). Results showed the Chao, Shannon, and Richness indexes were significantly positively correlated with total nitrogen content, whereas the Simpson index was significantly negatively correlated with water content. Apart from these associations, no significant linear regression relationships between α-diversity indexes and other physicochemical properties were detected.
3.3.2. Microbial β-Diversity
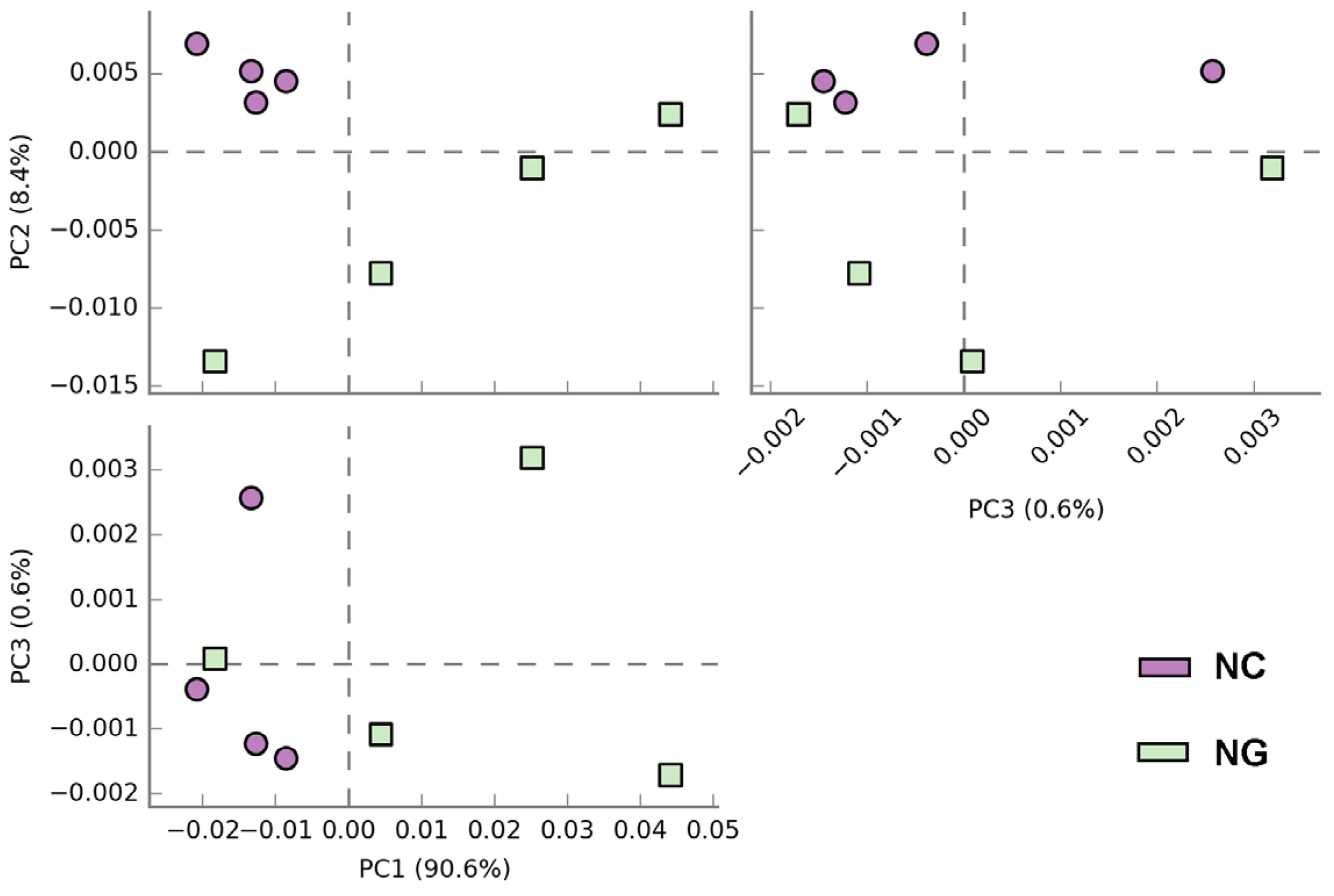
To further clarify the differences in community species composition between the two types of soil, PCoA analysis was used to determine the similarity of microbial species composition in rhizosphere and non-rhizosphere soils. The results in Figure 4 showed that principal coordinates (PC) 1, 2, and 3 had an overall explanation of 99.6%, with PC1 contributing 90.6%, indicating that the differences in microorganisms between rhizosphere and non-rhizosphere soil were mainly associated with the first principal coordinates. The PCoA plot showed that samples from non-rhizosphere soils tend to be closely clustered, indicating relatively similar community composition, whereas rhizosphere soil samples showed a more scattered distribution, indicating a degree of variation. The rhizosphere group (NG) and non-rhizosphere group (NC) had a large difference in distribution distance, indicating that there were large differences in microbial community composition between the two types of soil of Poa alpigena L.
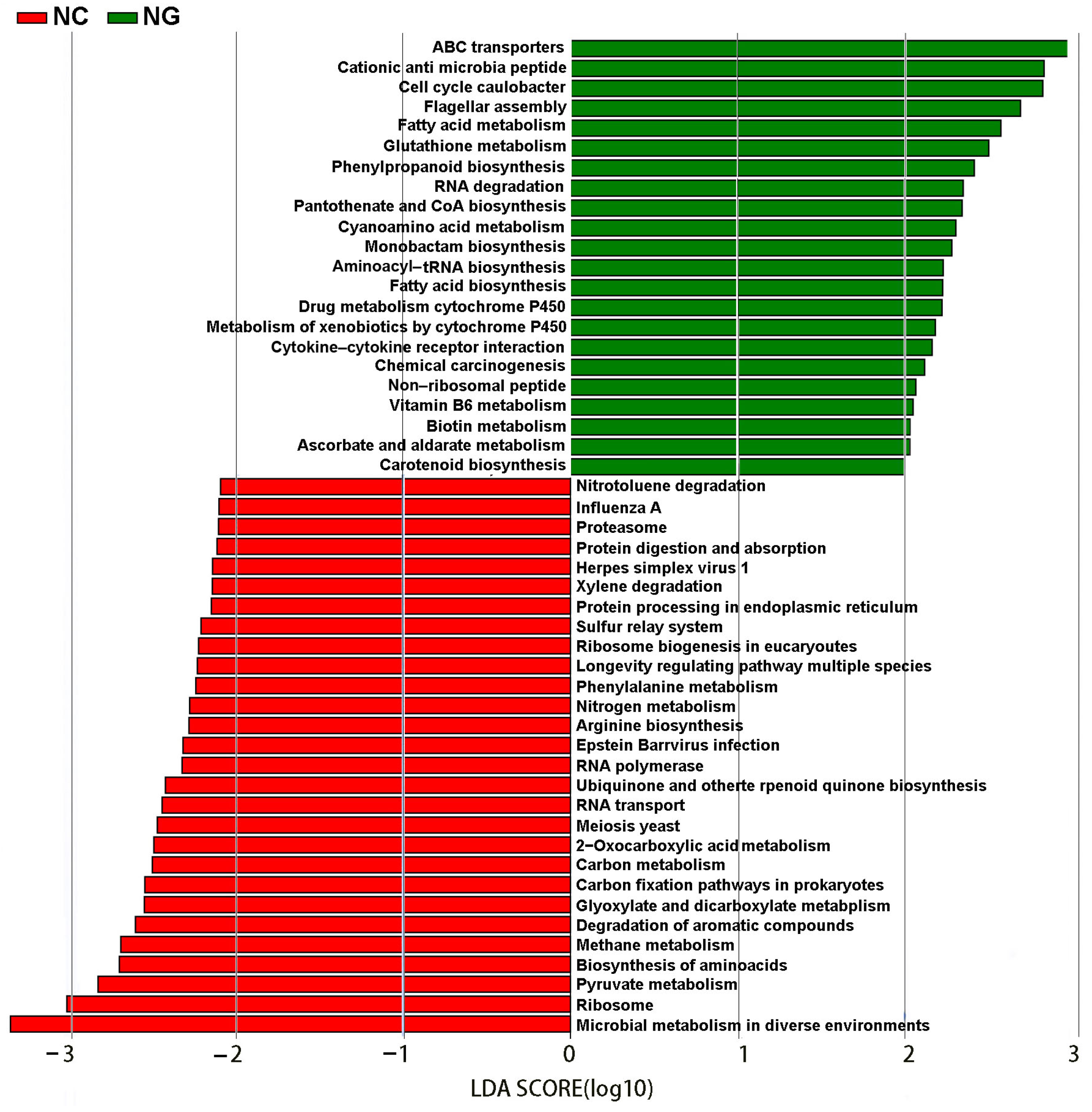
3.4. Differences in Metabolic Pathways between Rhizosphere and Non-Rhizosphere Soil Microorganisms
On the basis of LEfSe analysis of rhizosphere and non-rhizosphere microbial metabolic pathways, 51 pathways differing between the two type soils were identified (Figure 5), of which, 23 and 28 differential metabolic pathways were present in the rhizosphere and non-rhizosphere groups, respectively. Pathways associated with ATP-binding cassette (ABC) transporters, cationic antimicrobial peptide resistance, cell cycle-Caulobacter, fatty acid metabolism, glutathione metabolism, phenylpropanoid biosynthesis, RNA degradation, pantothenate and CoA biosynthesis, drug metabolism cytochrome P450, and metabolism ọf xenobiọtics by cytochrome P450 were significantly enriched in the rhizosphere group. Pathways associated with microbial metabolism, ribosome, pyruvate metabolism, biosynthesis of amino acids, methane metabolism, and degradation of aromatic compounds were significantly enriched in the non-rhizosphere group.
3.5. Correlation of Microorganisms of Poa alpigena L. Soils on Bird Island
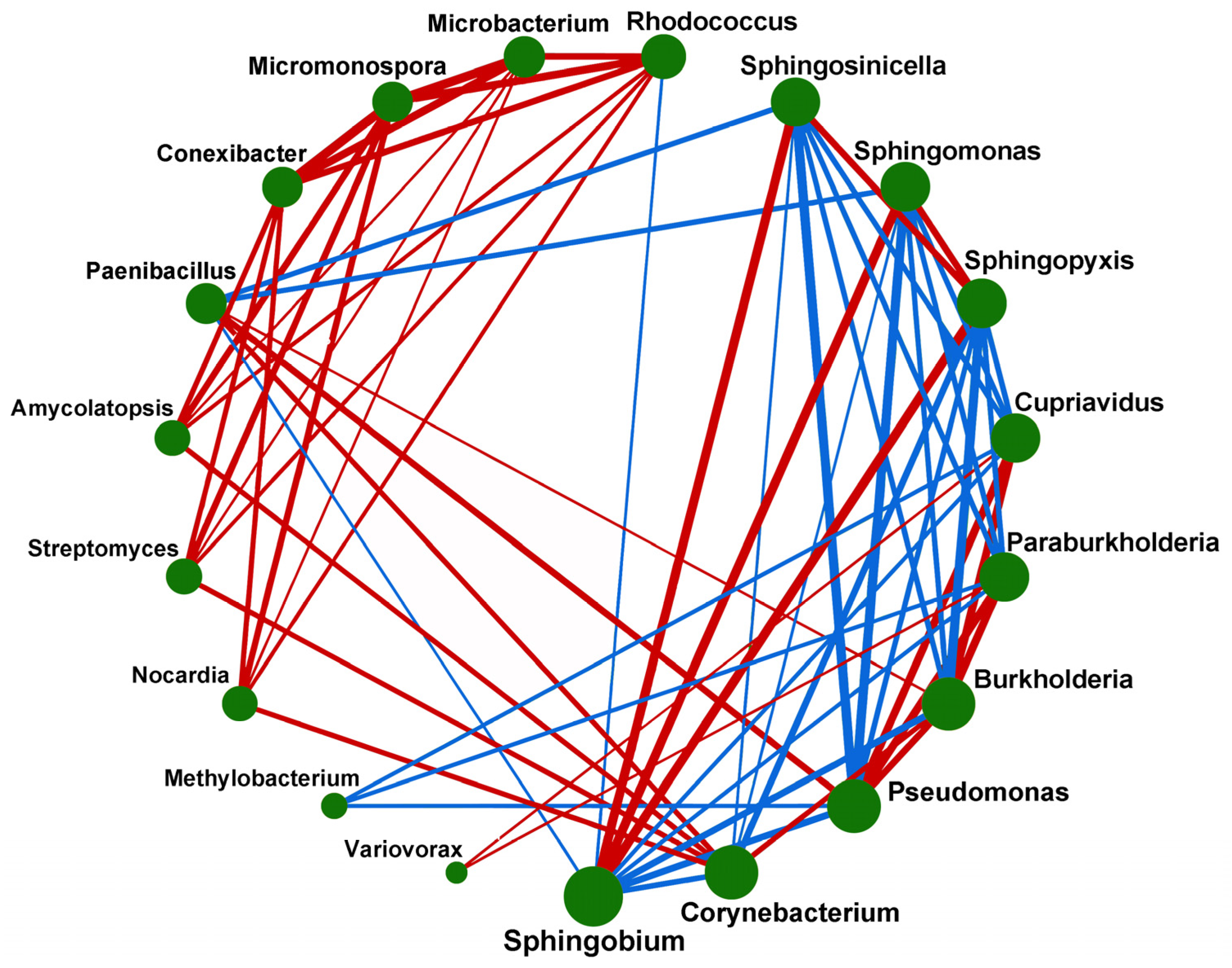
To examine the interrelationships among different microorganisms, the correlation analysis of microorganisms at the genus level was carried out in this study (Figure 6). The red and blue connecting lines, respectively, indicated the facilitatory and inhibitory relationships between the two connected genera, and the thickness of the lines indicated the absolute value of the correlation coefficient. The more the genera were related, the larger the green circle was. As shown in Figure 6, Sphingobium, Corynebacterium, and Pseudomonas had the most related genera, whereas Nocardia, Methylobacterium, and Variovorax had the fewest related genera. There were mutual facilitatory relationships between Burkholderia and Paraburkholderia, Burkholderia and Cupriavidus, and Cupriavidus and Variovorax. There were mutual inhibitory relationships between Burkholderia and Sphingopyxis, Pseudomonas and Sphingomonas, and Pseudomonas and Sphingosinicella. The facilitatory and inhibitory relationships between the different microorganisms indicated that different microorganisms interacted closely with each other and jointly influenced the metabolic process in the soil. However, the mechanisms underlying these microbial interactions need to be further studied.
4. Discussion
Although the microbial composition of rhizosphere and non-rhizosphere soils was essentially the same, there were notable differences in the relative abundance of various microorganisms at different taxonomic levels. At the phylum level, in terms of relative abundance, the dominant phyla in both rhizosphere and non-rhizosphere soils were Proteobacteria and Actinobacteria, which were consistent with the findings for soil dominant microorganisms in other types of habitats, particularly alpine soils. The results indicated that most of the dominant groups in soils were bacteria; among which, Proteobacteria and Actinobacteria were predominant, reflecting their broader ecological width and superior environmental adaptability [17,18]. However, there were significant differences in the relative abundance and the dominant groups at the lower levels of classification. The relative abundance of Proteobacteria in rhizosphere soil was significantly higher than that in non-rhizosphere soil, which may be attributable to the lower pH of rhizosphere soil that would provide an environment more conducive to the growth of Proteobacteria [19,20]. In the fungi, Ascomycota and Basidiomycota were identified as the predominant phyla. These fungi play important roles as decomposers in soil, wherein they, respectively, degrade the complex organic matter and lignocellulose from plant residues [21,22]. The relative abundances of Ascomycota and Basidiomycota in the non-rhizosphere soil of Poa alpigena L. were significantly higher than those in the rhizosphere soil, which might be related to the presence of a large number of dead leaf litter of Poa alpigena L. in the non-rhizosphere zone that needs to be degraded.
Streptomyces are widely distributed in extreme environments, including soils with high salinity, and they can produce important antibiotics and become an important medicinal resource [23]. At the genus level, Streptomyces has the highest abundance in both rhizosphere and non-rhizosphere soil, but the relative abundance of Streptomyces in non-rhizosphere soil was significantly higher than that in rhizosphere soil, probably because the non-rhizosphere soil environment around Poa alpigena L. is alkaline and the pH is greater than rhizosphere soil, which provided a more suitable growth environment for these bacteria. This finding provided data support for the exploitation of Streptomyces resource bank.
Stamp-based analysis of rhizosphere and non-rhizosphere differential microorganisms showed that the relative abundance of Candidate division NC10 was significantly lower (p < 0.05) in the rhizosphere soil than that in the non-rhizosphere group (Figure S2). These bacteria are involved in nitrite-dependent anaerobic methane oxidation (N-DAMO), which is associated with simultaneous soil denitrification and methane removal processes, and can mitigate the greenhouse effect [24]. The total nitrogen content in the non-rhizosphere soil was significantly higher than that in the rhizosphere soil, providing an abundant source of nitrogen for promoting the growth and reproduction of Candidate division NC10, as well as enriching the microbial nitrogen metabolism and methane metabolism pathways. The results of soil microbial community structure composition indicated that the diversity of bacteria was significantly higher than that of fungi in the soil of Poa alpigena L. in the Bird Island ecosystem on Qinghai Lake, which is consistent with the results of rhizosphere microbial studies in other soil ecosystems [25].
Diversity analysis of soil microorganisms showed that the microbial richness and diversity in non-rhizosphere soil of Poa alpigena L. were significantly higher than those in rhizosphere soil. This lower diversity in rhizosphere soil was assumed to reflect the production of specific root secretions, which were highly selective for certain soil microflora and less conducive to the proliferation of others [6,26]. Soil physicochemical properties were influenced to varying extents by the combined effects of plant root secretions and microbial metabolism, and in turn, soil total nitrogen and water content were important factors determining the diversity of soil microbial communities [15,27]. In this study, soil water content and total nitrogen content were significantly different in the rhizosphere and non-rhizosphere soil, which could significantly affect microbial diversity and richness. Low water content in rhizosphere soils inhibited microbial growth and activity, and low nitrogen content could not adequately meet the requirements for microbial growth and development, thus leading to reduced microbial diversity in rhizosphere soils [15,27,28]. There were differences between rhizosphere and non-rhizosphere soil pH of Poa alpigena L., and this difference may be because Poa alpigena L. roots improved rhizosphere nutrients by lowering rhizosphere pH, which facilitated the effective utilization of soil nutrients by Poa alpigena L. in a fragile environment, thus affecting the difference in microbial community structure and diversity [29,30].
Differences in the soil environment can variously influence microbial metabolic activities, and soil microorganisms were constantly adjusting metabolic pathways to adapt to environmental changes [31]. The large differences in the enriched pathways of rhizosphere and non-rhizosphere soil microbial communities indicated that soil microorganisms of Poa alpigena L. have adapted their metabolic pathways to some extent to adapt to the environment of the rhizosphere and non-rhizosphere areas, respectively. Among the metabolic pathways significantly enriched in rhizosphere soil microorganisms, in addition to certain basic metabolic pathways associated with normal life activities, an enrichment of pathways involving an ABC transporter, drug metabolism cytochrome P450, and metabolism xenobiotics by cytochrome P450 was detected. ABC transporter proteins can assist bacteria in hydrolyzing ATP to take up various nutrients from the environment. Similarly, Xu et al. found that the activity of ABC transporter proteins and their metabolic pathways were enhanced in sorghum rhizosphere soil microorganisms under drought conditions [32]. The soils of Bird Island are typically sandy with poor water holding capacity, and the enrichment of the ABC transporter pathway in rhizosphere soil microorganisms indicated that plants required larger amounts of ABC transporter proteins to facilitate the uptake of nutrients from the soil [33,34].
Drug and xenobiotic metabolisms are among the metabolic pathways associated with cytochrome P450, a major component of phase I biotransformation enzymes in organisms that catalyze the degradation and conversion of exogenous and endogenous substances, thereby enhancing resistance to exogenous stresses [35]. Studies on CYP450 mainly focused on humans, plants, and animals, and this study found an enrichment of CYP450-related metabolic pathways in rhizosphere microorganisms, suggesting that microorganisms in Poa alpigena L. roots may be stressed by exogenous pollutants, and CYP450 gene-related expression of microorganisms also had potential to indicate environmental changes [35,36,37].
Metabolic pathways related to gene expressions, such as RNA polymerase, RNA transport, ribosome, and proteasome were found to be significantly enriched in non- rhizosphere soil microorganisms, which indicated that gene expression is stronger in microorganisms in non-rhizosphere soil environments. This enhanced enrichment indicated that microorganisms in a root-free environment respond directly to external environmental stresses, which entailed an increased metabolic rate to facilitate environment adaptation, thus the respiration-related pathways associated with ubiquinone and other terpenoid-quinone biosynthesis were also significantly enriched to increase resistance [38].
Pathways associated with methane metabolism and degradation of aromatic compounds were also significantly enriched in the non-rhizosphere soil microorganisms. In non-rhizosphere soils, the enrichment of the methane metabolism pathway was associated with a higher relative abundance of Candidate division NC10 involved in N-DAMO [39]. The enrichment of the pathway associated with the degradation of aromatic compounds indicated that microorganisms in this region have evolved the ability to use aromatic compounds as a potential source of carbon and energy. By degrading these compounds, these microbes not only obtain energy for survival but also contribute to eliminating potentially toxic aromatic compounds from the environment. The significant enrichment of this metabolic pathway provided evidence to indicate that there might be aromatic compound pollution in the soil environment and provided an early warning of soil ecological pollution in this region [40].
The microbial community structure is complex, so it is inevitable and reasonable for microorganisms to form a mutual facilitatory or inhibitory relationship, and this relationship is usually to compete for energy and materials, so as to determine species’ presence and relative abundance to a certain extent [41]. In this study, facilitatory interactions among Cupriavidus, Burkholderia and Variovorax were detected. Cupriavidus is highly resistant to heavy metals, whereas Burkholderia is one kind of microorganism beneficial for plant growth and adaptation to the environment, and Variovorax maintains directional root development by controlling plant auxin and ethylene levels. Thus, the mutual facilitatory relationships among Cupriavidus, Burkholderia, and Variovorax were beneficial to improving the root development and adaptability of Poa alpigena L. to environmental stress, and providing support for land restoration in the Qinghai Lake basin [42,43,44].
5. Conclusions
In this study, through the characterization of the soil microbial communities in the rhizosphere and non-rhizosphere soils of Poa alpigena grassland on Bird Island in the Qinghai Lake basin, the following conclusions were obtained.
(1) The dominant bacteria in the rhizosphere and non-rhizosphere soils of Poa alpigena L. on Bird Island were Proteobacteria and Actinobacteria, whereas the dominant fungi were Ascomycota and Basidiomycota. The rhizosphere of Poa alpigena L. was more selective for soil microflora, resulting in lower microbial richness and diversity in rhizosphere soil than those in non-rhizosphere soil. Different environmental factors, notably soil water content and total nitrogen, produced varying effects on soil microbial diversity. (2) The results of the KEGG pathway enrichment analysis indicated that rhizosphere microorganisms required more ABC transporters and cytochrome P450-related metabolism to obtain energy for growth and to cope with exogenous environmental stress. The significant enrichment of gene expression-related pathways in non-rhizosphere soil microorganisms, as well as pathways associated with methane metabolism and degradation of aromatic compounds, indicated that the soil microbial community associated with the Poa alpigena L. community played an important role in the oxidation of methane and regulation of the greenhouse effect in the Qinghai Lake basin. Pathway analysis also indicated that there may be some degree of risk of aromatic compound contamination in Poa alpigena L. soils on Bird Island, which can be analyzed in conjunction with soil contaminant detection. (3) Correlation analysis revealed multiple facilitatory and inhibitory relationships among the microbial community in Poa alpigena L. soil. However, the mechanisms underlying these microbial interactions are in need of further study. The results of this study can provide a scientific basis for the control of the greenhouse effect in the Qinghai Lake basin, promote the construction of a regional microbial resource bank, and contribute to the detection of soil pollution.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/w15020239/s1, Figure S1: The relative abundances of microorganisms at the class (a), order (b), family (c), and species (d) levels in rhizosphere (NG) and non-rhizosphere (NC) soil; Figure S2: Scale plot of the characteristic sequence of Candidate division NC10.
Author Contributions
Conceptualization, L.L. and H.W.; methodology, L.L. and H.W.; software, Y.C. and H.W.; validation, L.Q. and K.C.; formal analysis, L.L.; investigation, Z.C. and H.W.; resources L.L.; data curation, L.L. and H.W.; writing—original draft preparation, L.L.; writing—review and editing, L.L. and H.W.; visualization, H.C and H.W.; supervision, H.W. and K.C.; project administration, L.L. and H.W.; funding acquisition, L.L., H.W. and K.C. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the Natural Science Foundation of Anhui Province (grant No. 2008085QC103); The National Natural Science Foundation of P.R. China (grant No. 41807322); the Natural Science Research Project of Anhui Provincial Colleges and Universities (grant No. KJ2021A0924, KJ2020A0116); and the Second Tibetan Plateau Scientific Expedition and Research Program (grant No. 2019QZKK0405).
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data have been uploaded to NCBI.
Acknowledgments
Thanks to the National Positioning Observation and Research Station of the Qinghai Lake Wetland Ecosystem in Qinghai Province for providing technical support, to all the authors of this article, and to the editor and anonymous reviewers for their constructive comments and suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, N.N.; Sun, G.; Liang, J.; Wang, E.T.; Shi, C.G.; He, J.; Hu, X.; Zhao, C.Z.; Wu, N. Response of ammonium oxidizers to the application of nitrogen fertilizer in an alpine meadow on the Qinghai-Tibetan Plateau. Appl. Soil Ecol. 2018, 124, 266–274. [Google Scholar] [CrossRef]
- Zhou, T.Q.; Kong, W.D.; Chen, H. Advances in microbial research on Tibetan grassland soil. Chin. J. Ecol. 2022. Available online: https://kns.cnki.net/kcms/detail/21.1148.Q.20220520.1948.043.html (accessed on 15 November 2022). (In Chinese).
- Liu, J.B.; Kong, W.D.; Zhang, G.S.; Khan, A.; Guo, G.X.; Zhu, C.M.; Wei, X.J.; Kang, S.C.; Morgan-Kiss, R.M. Diversity and succession of autotrophic microbial community in high elevation soils along deglaciation chronosequence. FEMS Microbiol. Ecol. 2016, 92, fiw160. [Google Scholar] [CrossRef] [Green Version]
- Marilley, L.; Vogt, G.; Blanc, M.; Aragno, M. Bacterial diversity in the bulk soil and rhizosphere fractions of Lolium perenne and Trifolium repens as revealed by PCR restriction analysis of 16S rDNA. Plant Soil 1998, 198, 219–224. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-Growth-Promoting Rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Grayston, S.J.; Wang, S.Q.; Campbell, C.D.; Anthony, C.E. Selective influence of plant species on microbial diversity in the rhizosphere. Soil Biol. Biochem. 1998, 30, 369–378. [Google Scholar] [CrossRef]
- Jia, J.J.; Wang, Y.F.; Lu, Y.; Sun, K.; Lyu, S.D.; Gao, Y. Driving mechanisms of gross primary productivity geographical patterns for Qinghai-Tibet Plateau lake systems. Sci. Total Environ. 2021, 791, 148286–148299. [Google Scholar] [CrossRef]
- Zhai, W.T.; Chen, D.D.; Li, Q.; Zhao, L.; Liu, Z.; Xu, S.X.; Dong, Q.M.; Zhao, X.Q. Effect of grazing intensity on carbon metabolic characteristics of soil microbial communities in an alpine steppe in the regions around Qinghai Lake. Chin. J. Appl. Environ. Biol. 2017, 23, 685–692. (In Chinese) [Google Scholar]
- Wang, X.Y.; Li, Y.Q.; Gong, X.W.; Niu, Y.Y.; Chen, Y.P.; Shi, X.P.; Li, W.; Liu, J. Changes of soil organic carbon stocks from the 1980s to 2018 in northern China’s agro-pastoral ecotone. Catena 2020, 194, 104722. [Google Scholar] [CrossRef]
- Shi, H.X.; Hou, X.Y.; Shi, S.L.; Wu, X.H.; Yang, T.T.; Li, P. Poa alpigena response traits affected by grazing and enclosuresin an alpine meadow on the Qinghai-Tibet Plateau. Acta Ecol. Sin. 2016, 36, 3601–3608. (In Chinese) [Google Scholar]
- Wei, L.N.; Zhang, C.P.; Dong, Q.M.; Yu, Y.; Yang, X.X. Characterization of the complete chloroplast genome of Poa pratensis L. cv. Qinghai (Gramineae). Mitochondrial DNA Part B Resour. 2020, 5, 532–533. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.J.; Ni, Y.; Liao, L.X.; Xiao, Y.; Guo, Y.J. Poa pratensis ECERIFERUM1 (PpCER1) is involved in wax alkane biosynthesis and plant drought tolerance. Plant Physiol. Biochem. 2021, 159, 312–321. [Google Scholar] [CrossRef]
- Dong, W.K.; Ma, X.; Jiang, H.Y.; Zhao, C.X.; Ma, H.L. Physiological and transcriptome analysis of Poa pratensis var. anceps cv. Qinghai in response to cold stress. BMC Plant Biol. 2020, 20, 362. [Google Scholar] [CrossRef]
- Riley, D.; Barber, S.A. Effect of ammonium and nitrate fertilization on phosphorus uptake as related to root-Induced pH changes at the root-soil interface. Soil Sci. Soc. Amer. Proc. 1971, 35, 301–306. [Google Scholar] [CrossRef]
- Liu, F.D.; Mo, X.; Kong, W.J.; Song, Y. Soil bacterial diversity, structure, and function of Suaeda salsa in rhizosphere and non-rhizosphere soils in various habitats in the Yellow River Delta, China. Sci. Total Environ. 2020, 740, 140144. [Google Scholar] [CrossRef]
- Han, G.M.; Song, F.Q.; Zhang, Z.J.; Ni, W.; He, S.E.; Tian, X.J. An economic and efficient method for further purification of crude DNA extracted from forest soils. J. Forestry Res. 2010, 21, 246–250. [Google Scholar] [CrossRef]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.M.; Provin, T.L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Sheng, H.Y.; Luo, S.S.; Hu, Y.M.; Yu, L.L. Characteristics of prokaryotic microbial community structure and molecular ecological network in four habitat soils around Lake Qinghai. Ecol. Environ. Sci. 2021, 30, 1393–1403. (In Chinese) [Google Scholar]
- Yang, H.J.; Wang, Q.; Wan, Z.X.; Zhang, Z.Y.; Chen, D.D.; Tan, J. Structure and diversity of microbial communities in the rhizosphere and non-rhizosphere soil in areas with invasive Solidago canadensis L. J. Biosaf. 2021, 30, 235–243. (In Chinese) [Google Scholar]
- Zhang, P.; Cui, Z.Y.; Guo, M.Q.; Xi, R.C. Characteristics of the soil microbial community in the forestland of Camellia oleifera. PeerJ 2020, 8, 9117. [Google Scholar] [CrossRef]
- Frey, S.D.; Knorr, M.; Parrent, J.L.; Simpson, R.T. Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. For. Ecol. Manag. 2004, 196, 159–171. [Google Scholar] [CrossRef]
- Beimforde, C.; Feldberg, K.; Nylinder, S.; Rikkinen, J.; Tuovila, H.; Dörfelt, H.; Gube, M.; Jackson, D.J.; Reitner, J.; Seyfullah, L.J.; et al. Estimating the Phanerozoic history of the Ascomycota lineages: Combining fossil and molecular data. Mol. Phylogenet Evol. 2014, 78, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cao, Y.R.; Wiese, J.; Tang, S.K.; Xu, L.H.; Imhoff, J.F.; Jiang, C.L. Streptomyces sparsus sp. nov., isolated from a saline and alkaline soil. Int. J. Syst. Evol Microbiol. 2011, 61, 1601–1605. [Google Scholar] [CrossRef] [Green Version]
- Harbison, A.B.; Garson, M.A.; Lamit, L.J.; Basiliko, N.; Bräuer, S.L. A novel isolate and widespread abundance of the candidate alphaproteobacterial order (Ellin 329), in southern Appalachian peatlands. FEMS Microbiol. Lett. 2016, 363, fnw151. [Google Scholar] [CrossRef] [Green Version]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Marilley, L.; Aragno, M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Loliumperenne and Trifolium repens roots. Appl. Soil Ecol. 1999, 13, 127–136. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, B.W.; Sun, R.D.; Zhang, Y.Y.; Song, J.Y.; Zhang, G.J.; Xu, L.; Mu, R.L. Characteristics of culturable microbial communities in rhizosphere /nonrhizosphere soil of Ligularia virgaurea in alpine meadow elevation gradient. Acta Ecol. Sin. 2021, 41, 4853–4863. (In Chinese) [Google Scholar]
- Zhang, K.; Bao, W.K.; Yang, B.; Hu, B. The effects of understory vegetation on soil microbial community composition and structure. Chin. J. Appl. Environ. Biol. 2017, 23, 1178–1184. (In Chinese) [Google Scholar]
- Yang, Y.; Liu, B.R. Distribution of soil nutrient and microbial biomass in rhizosphere versus non-rhizosphere area of different plant species in desertified steppe. Acta Ecol. Sin. 2015, 35, 7562–7570. [Google Scholar]
- Wang, Z.H.; Jiang, X.J. Contrasting responses of the microbial community structure and functional traits to soil pH in Purple Soils. Environ. Sci. 2022, 43, 3876–3883. [Google Scholar]
- Palit, K.; Rath, S.; Chatterjee, S.; Das, S. Microbial diversity and ecological interactions of microorganisms in the mangrove ecosystem: Threats, vulnerability, and adaptations. Environ. Sci. Pollut. Res. 2022, 29, 32467–32512. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 4284–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenstein, K.; Dawson, R.J.P.; Locher, K.P. Struct and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T. Structural dynamics of ABC transporters: Molecular simulation studies. Biochem. Soc. Trans. 2021, 49, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, X.; Wang, H.; Liu, T.; Cheng, J.; Jiang, H. Discovery and modification of cytochrome P450 for plant natural products biosynthesis. Synth. Syst. Biotechnol. 2020, 5, 187–199. [Google Scholar] [CrossRef]
- Waring, R.H. Waring Cytochrome P450: Genotype to phenotype. Xenobiotica 2020, 50, 9–18. [Google Scholar] [CrossRef]
- Murayama, N.; Yamazaki, H. Metabolic activation and deactivation of dietary-derived coumarin mediated by cytochrome P450 enzymes in rat and human liver preparations. J. Toxicol. Sci. 2021, 46, 371–378. [Google Scholar] [CrossRef]
- Nie, J.T.; Wang, H.S.; Zhang, W.L.; Teng, X.; Yu, C.; Cai, R.; Wu, G. Characterization of lncRNAs and mRNAs involved in powdery mildew resistance in Cucumber. Phytopathology 2021, 111, 1613–1624. [Google Scholar] [CrossRef]
- Ettwig, K.F.; Butler, M.K.; Le, P.D.; Pelletier, E.; Mangenot, S.; Kuypers, M.M.; Schreiber, F.; Dutilh, B.E.; Zedelius, J.; de Beer, D. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 2010, 464, 543–548. [Google Scholar] [CrossRef]
- Phale, P.S.; Basu, A.; Majhi, P.D.; Deveryshetty, J.; Vamsee-Krishna, C.; Shrivastava, R. Metabolic diversity in bacterial degradation of aromatic compounds. OMICS J. Integr. Biol. 2007, 11, 252–279. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.R.; Huang, Y.C.; Yang, X.R.; Xue, W.J.; Zhang, X.; Zhang, Y.H.; Pang, J.; Liu, Y.M.; Liu, Z.Q. Burkholderia sp. Y4 inhibits cadmium accumulation in rice by increasing essential nutrient uptake and preferentially absorbing cadmium. Chemosphere 2020, 252, 126603. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.Y.; Lv, Y.K.; Liu, Y.X.; Ren, R.P. Removal of nitrogen by heterotrophic nitrification-aerobic denitrification of a novel metal resistant bacterium Cupriavidus sp. S1. Bioresour. Technol. 2016, 220, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Salas-González, I.; Castrillo, G.; Conway, J.M.; Law, T.F.; Teixeira, P.J.P.L.; Wilson, E.D.; Fitzpatrick, C.R.; Jones, C.D.; Dangl, J.L. A single bacterial genus maintains root growth in a complex microbiome. Nature 2020, 587, 103–133. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) The relative abundances of microorganisms at the phylum level in rhizosphere (NG) and non-rhizosphere (NC) soil and (b) the relative abundances of microorganisms at the genus level in rhizosphere (NG) and non-rhizosphere (NC) soil.
Figure 1.
(a) The relative abundances of microorganisms at the phylum level in rhizosphere (NG) and non-rhizosphere (NC) soil and (b) the relative abundances of microorganisms at the genus level in rhizosphere (NG) and non-rhizosphere (NC) soil.

Figure 2.
(a) Classification level discriminant analysis (LDA) of soil microbial communities by LEfSe analysis and (b) a cladogram of soil microbial communities by LEfSe analysis.
Figure 2.
(a) Classification level discriminant analysis (LDA) of soil microbial communities by LEfSe analysis and (b) a cladogram of soil microbial communities by LEfSe analysis.

Figure 3.
A simple linear regression analysis between soil water content, total nitrogen, and α-diversity indexes. Note: * indicated the linear regression was significant (p < 0.05), indicators for which regressions were not significant were not shown.
Figure 3.
A simple linear regression analysis between soil water content, total nitrogen, and α-diversity indexes. Note: * indicated the linear regression was significant (p < 0.05), indicators for which regressions were not significant were not shown.

Figure 4.
Principal coordinates analysis (PCoA) of a microorganism community in rhizosphere and non-rhizosphere soil.
Figure 4.
Principal coordinates analysis (PCoA) of a microorganism community in rhizosphere and non-rhizosphere soil.

Figure 5.
Classification level discriminant analysis (LDA) of the KEGG pathway by LEfSe analysis.

Figure 6.
Ecological networks of soil microbial community in soil. Note: The size of the green circle indicated the number of related genera. The more the genera were related, the larger the circle was. The red and blue connecting lines, respectively, indicated the facilitatory and inhibitory relationship between the two connected genera, and the thickness of the lines indicated the absolute value of the correlation coefficient.
Figure 6.
Ecological networks of soil microbial community in soil. Note: The size of the green circle indicated the number of related genera. The more the genera were related, the larger the circle was. The red and blue connecting lines, respectively, indicated the facilitatory and inhibitory relationship between the two connected genera, and the thickness of the lines indicated the absolute value of the correlation coefficient.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The physicochemical properties of soil.
| Soil Properties | NC | NG |
|---|---|---|
| Water content (%) | 0.24 ± 0.02 a | 0.15 ± 0.01 b |
| pH | 8.91 ± 0.17 a | 8.59 ± 0.30 a |
| Electronic conductivity (ms cm−1) | 0.05 ± 0.01 a | 0.06 ± 0.02 a |
| Total carbon (g kg−1) | 5.12 ± 0.40 a | 5.14 ± 0.45 a |
| Total nitrogen (g kg−1) | 0.42 ± 0.05 a | 0.26 ± 0.03 b |
Notes: Data are expressed as mean ± SD and the significance of different soils is marked with different letters (p < 0.05). NG is rhizosphere soil and NC is non-rhizosphere soil.
Table 2.
Microbial sequencing results of Poa alpigena L. soil on Bird Island in Qinghai Lake.
| Project Name | NC | NG |
|---|---|---|
| Number of clean reads (couple) | 36,702,067.5 ± 1074957.9 a | 36,367,431.5 ± 2157852.8 a |
| Unclassified reads (%) | 57.13 ± 0.72 a | 54.72 ± 1.55 b |
| Classified reads (%) | 42.88 ± 0.72 b | 45.28 ± 1.55 a |
| Chordate reads (%) | 6.23 ± 0.06 a | 6.20 ± 0.27 a |
| Microbial reads (%) | 36.50 ± 1.00 b | 38.94 ± 2.00 a |
| Bacterial reads (%) | 28.80 ± 0.54 a | 31.22 ± 1.91 a |
| Fungal reads (%) | 1.42 ± 0.04 a | 1.42 ± 0.06 a |
| Viral reads (%) | 0.16 ± 0.00 a | 0.16 ± 0.00 a |
| Protozoan reads (%) | 0.18 ± 0.00 a | 0.18 ± 0.01 a |
Notes: The percentages in the table refer to the corresponding sequence as a percentage of the total sequence. Data are expressed as mean ± SD and the significance of different soils is marked with different letters (p < 0.05). NG is the rhizosphere group and NC is the non-rhizosphere group.
Table 3.
The α-diversity indexes of microorganisms in rhizosphere (NG) and non-rhizosphere (NC) soil.
Table 3.
The α-diversity indexes of microorganisms in rhizosphere (NG) and non-rhizosphere (NC) soil.
| Alpha Diversity Indexes | NC | NG |
|---|---|---|
| Richness index | 5702.00 ± 2.31 a | 5654.75 ± 31.73 b |
| Chao 1 index | 5703.18 ± 2.28 a | 5655.98 ± 31.69 b |
| Shannon index | 7.78 ± 0.01 a | 7.73 ± 0.01 b |
| Simpson index | 1.10 × 10−3 ± 2.52 × 10−5 b | 1.15 × 10−3 ± 2.58 × 10−5 a |
Note: Data are expressed as mean ± SD and the significance of different soils is marked with different letters (p < 0.05). NG is rhizosphere soil and NC is non-rhizosphere soil.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, L.; Che, Z.; Cao, Y.; Qi, L.; Chen, K.; Wang, H. Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis. Water 2023, 15, 239. https://doi.org/10.3390/w15020239
AMA Style
Li L, Che Z, Cao Y, Qi L, Chen K, Wang H. Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis. Water. 2023; 15(2):239. https://doi.org/10.3390/w15020239
Chicago/Turabian StyleLi, Lingling, Zihan Che, Yanhong Cao, Lulu Qi, Kelong Chen, and Hengsheng Wang. 2023. "Analyzing the Soil Microbial Characteristics of Poa alpigena Lindm. on Bird Island in Qinghai Lake Based on Metagenomics Analysis" Water 15, no. 2: 239. https://doi.org/10.3390/w15020239
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.