
Comparative Analysis of Metagenomic (Amplicon and Shotgun) DNA Sequencing to Characterize Microbial Communities in Household On-Site Wastewater Treatment Systems
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
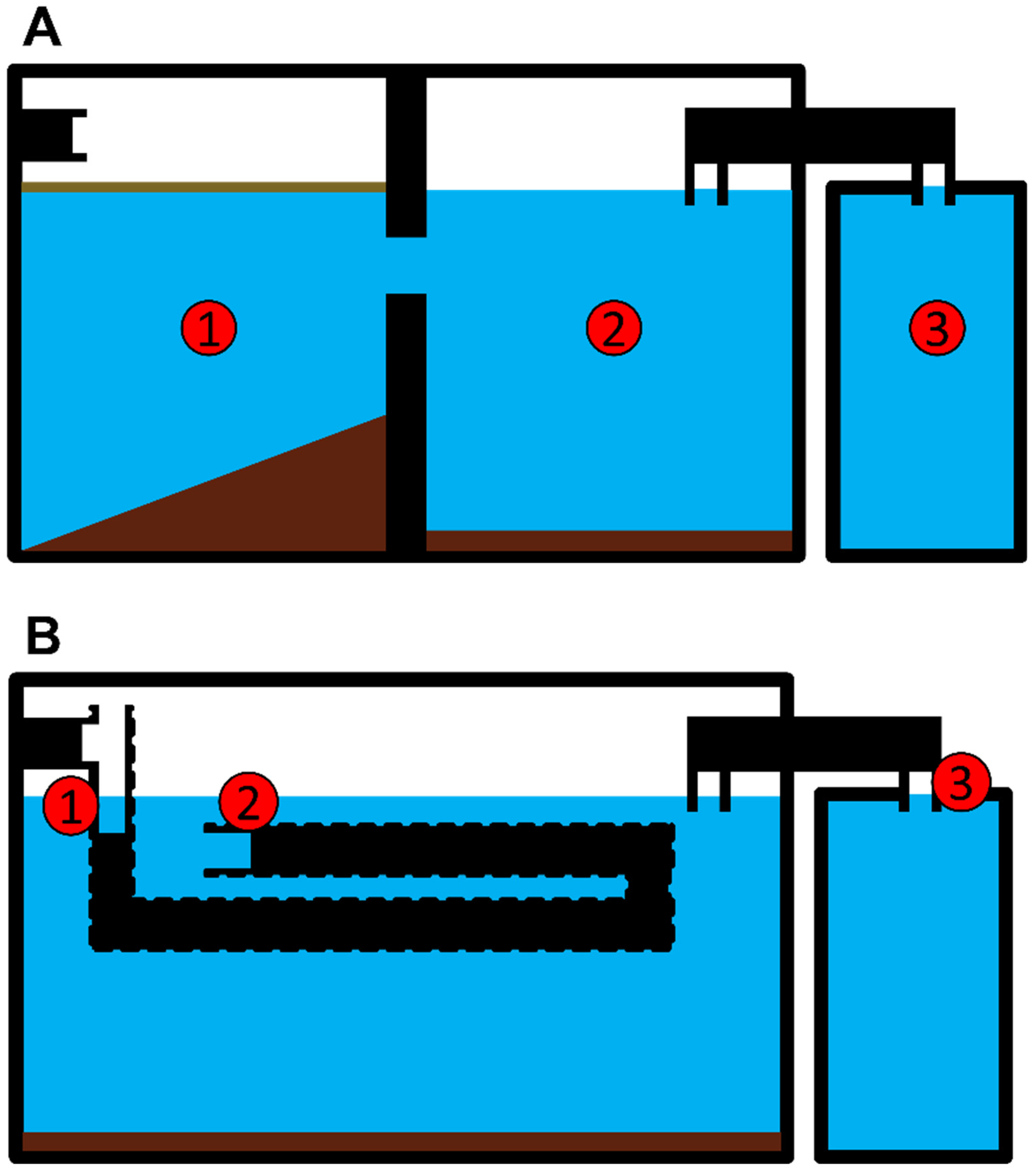
2.1. Sampling Site Description
2.2. Sampling Procedure
2.3. DNA Extraction
2.4. Shotgun Sequencing, Quality Control, and Classification
2.5. 16S Amplification, Sequencing, Quality Control, and Classification
2.6. Statistical Analysis
3. Results and Discussion
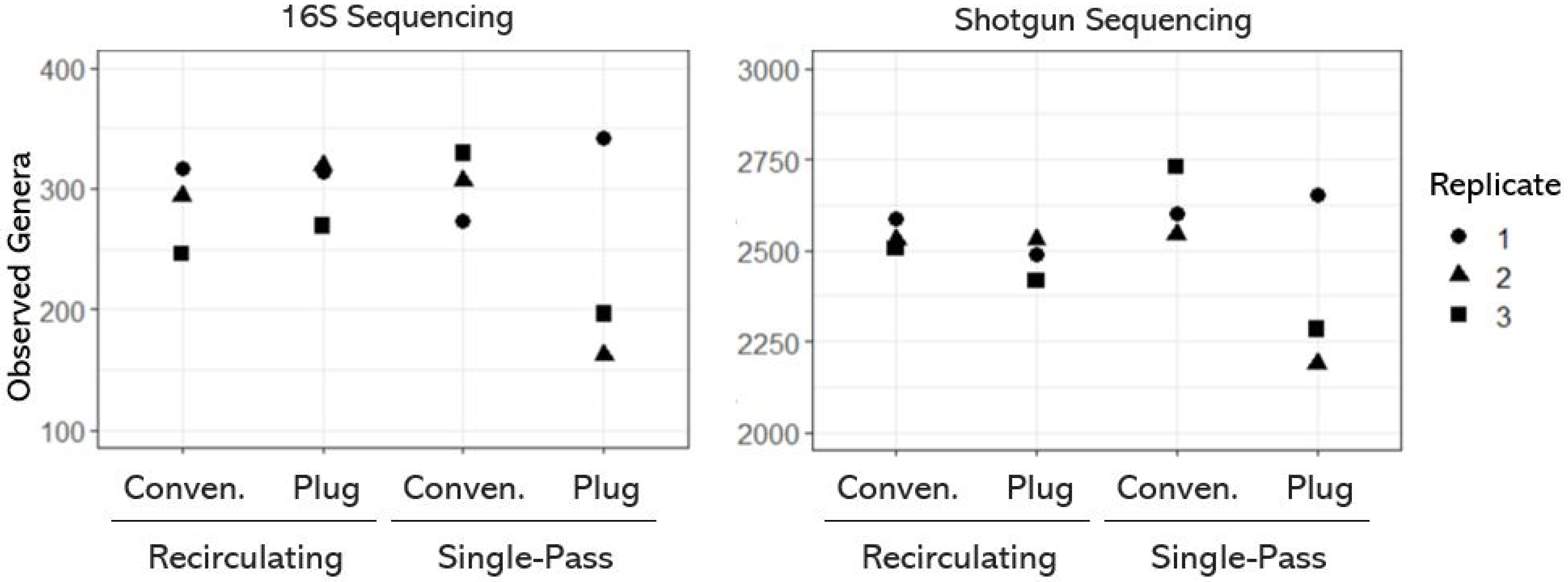
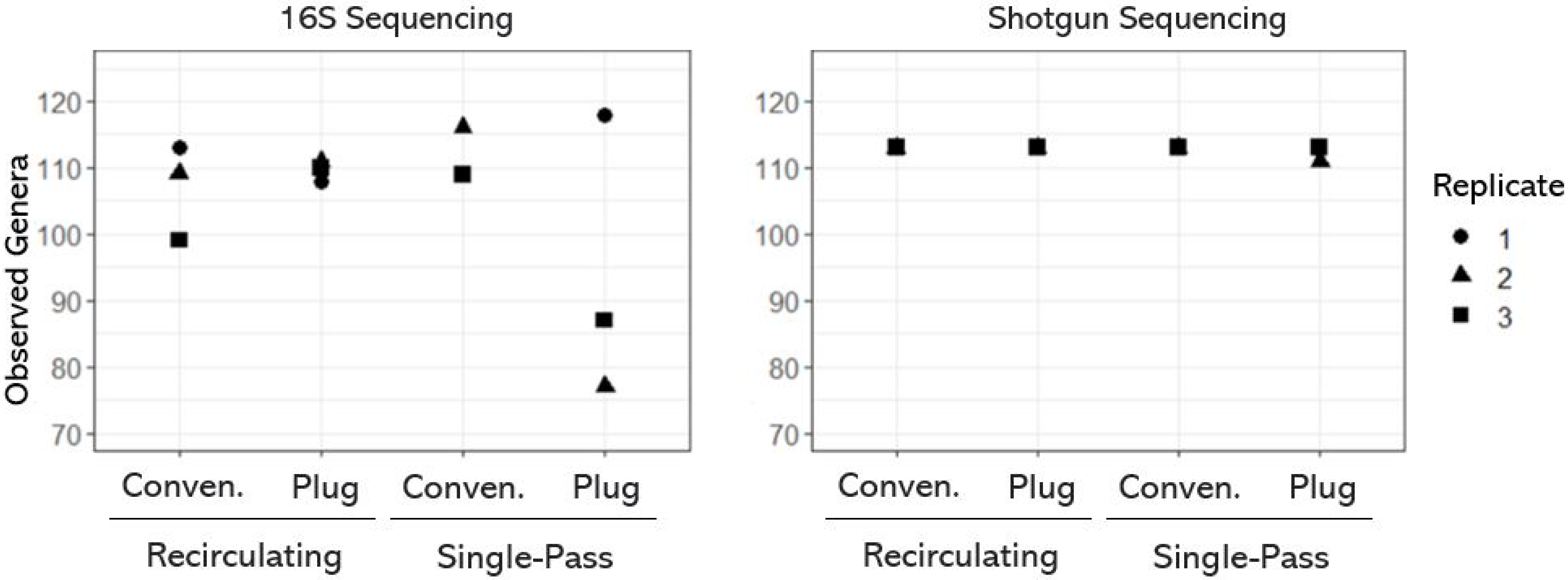
3.1. Taxonomic Diversity (or Richness) of OWTS Microbial Communities
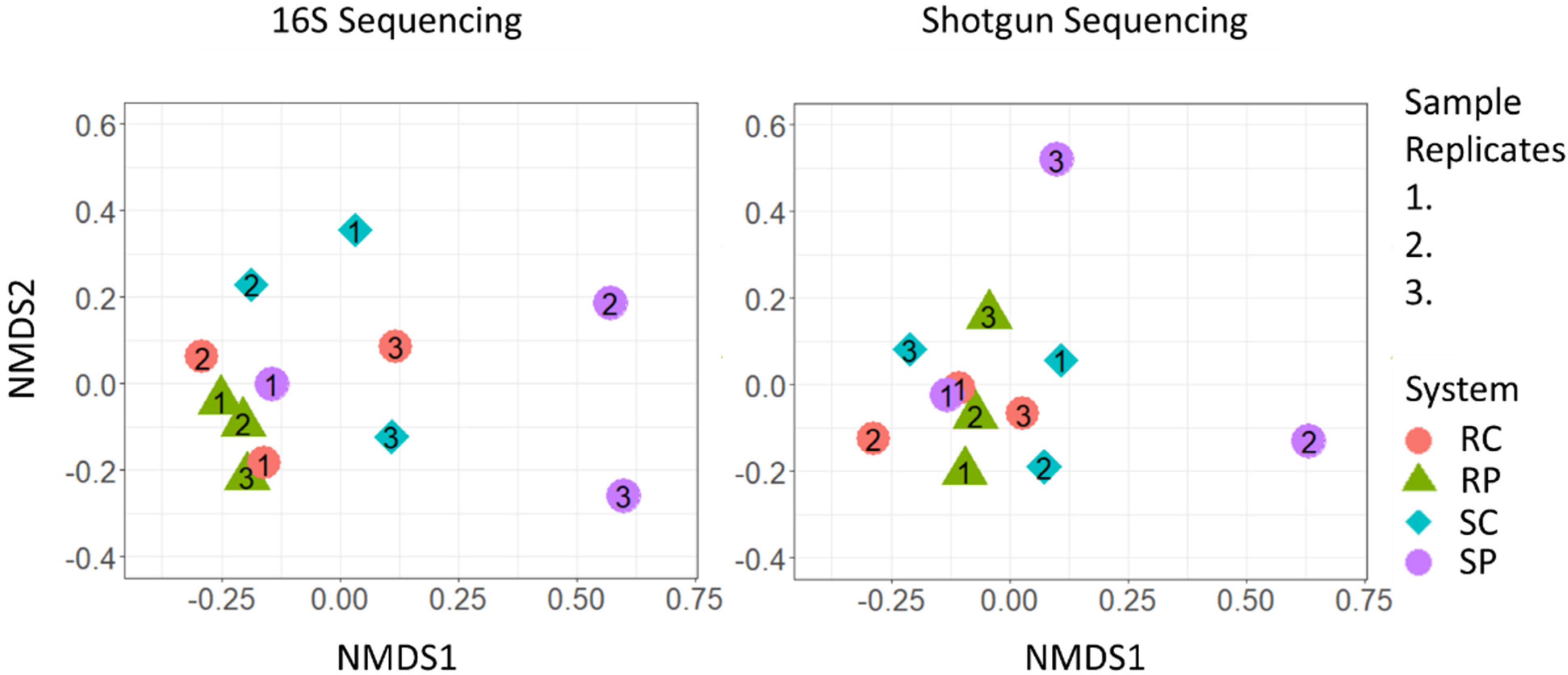
3.2. Community Differentiation and Core Microbiome
3.3. Differential Abundance
4. Conclusions
- The OWTSs designed with a recirculating flow system and plug-flow-type design contained the most variable taxonomic richness.
- Single-pass plug-flow-type OWTSs contained the most variable microbial communities between OWTSs.
- Desulfomicrobium was enriched in conventional OWTSs, whereas Simplicispira and Phenylobacterium were both enriched in single-pass OWTSs.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richards, S.; Withers, P.J.A.; Paterson, E.; McRoberts, C.W.; Stutter, M. Potential Tracers for Tracking Septic Tank Effluent Discharges in Watercourses. Environ. Pollut. 2017, 228, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Saleem, F.; Edge, T.A.; Schellhorn, H.E. Biological indicators for fecal pollution detection and source tracking: A review. Processes 2021, 9, 2058. [Google Scholar] [CrossRef]
- Appling, D.; Habteselassie, M.Y.; Radcliffe, D.; Bradshaw, J.K. Preliminary Study on the Effect of Wastewater Storage in Septic Tank on E. coli Concentration in Summer. Water 2013, 5, 1141–1151. [Google Scholar] [CrossRef]
- Schaider, L.A.; Rodgers, K.M.; Rudel, R.A. Review of Organic Wastewater Compound Concentrations and Removal in Onsite Wastewater Treatment Systems. Environ. Sci. Technol. 2017, 51, 7304–7317. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, B.E.; Herren, L.W.; Paule, A.L. Septic Systems Contribute to Nutrient Pollution and Harmful Algal Blooms in the St. Lucie Estuary, Southeast Florida, USA. Harmful Algae 2017, 70, 1–22. [Google Scholar] [CrossRef]
- Grattan, L.M.; Holobaugh, S.; Morris, J.G. Harmful Algal Blooms and Public Health. Harmful Algae 2016, 57, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Withers, P.J.A.; Jordan, P.; May, L.; Jarvie, H.P.; Deal, N.E. Do Septic Tank Systems Pose a Hidden Threat to Water Quality? Front. Ecol. Environ. 2014, 12, 123–130. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Krooneman, J.; Euverink, G.J.W. Strategies to boost anaerobic digestion performance of cow manure: Laboratory achievements and their full-scale application potential. Sci. Total Environ. 2021, 755, 142940. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, W.; Liu, C.; Zhang, R.; Liu, G. Mitigation of ammonia inhibition through bioaugmentation with different microorganisms during anaerobic digestion: Selection of strains and reactor performance evaluation. Water Res. 2019, 155, 214–224. [Google Scholar] [CrossRef]
- Fekih-Salem, R.; Daoud, Y.; Abdellatif, N.; Sari, T. A mathematical model of anaerobic digestion with syntrophic relationship, substrate inhibition, and distinct removal rates. SIAM J. Appl. Dyn. Syst. 2021, 20, 1621–1654. [Google Scholar] [CrossRef]
- Naphtali, J.; Chan, A.W.Y.Y.; Saleem, F.; Li, E.; Devries, J.; Schellhorn, H.E. Comparative Metagenomics of Anaerobic Digester Communities Reveals Sulfidogenic and Methanogenic Microbial Subgroups in Conventional and Plug Flow Residential Septic Tank Systems. Processes 2022, 10, 436. [Google Scholar] [CrossRef]
- Chen, J.; Wade, M.J.; Dolfing, J.; Soyer, O.S. Increasing Sulfate Levels Show a Differential Impact on Synthetic Communities Comprising Different Methanogens and a Sulfate Reducer. J. R. Soc. Interface 2019, 16, 20190129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar-Zepeda, A.; Godoy-Lozano, E.E.; Raggi, L.; Segovia, L.; Merino, E.; Gutiérrez-Rios, R.M.; Juarez, K.; Licea-Navarro, A.F.; Pardo-Lopez, L.; Sanchez-Flores, A. Analysis of Sequencing Strategies and Tools for Taxonomic Annotation: Defining Standards for Progressive Metagenomics. Sci. Rep. 2018, 8, 12034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas-Lloret, J.; Obón-Santacana, M.; Ibáñez-Sanz, G.; Guinó, E.; Pato, M.L.; Rodriguez-Moranta, F.; Mata, A.; García-Rodríguez, A.; Moreno, V.; Pimenoff, V.N. Gut Microbiome Diversity Detected by High-Coverage 16S and Shotgun Sequencing of Paired Stool and Solon Sample. Sci. Data 2020, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison Between 16s rRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; Desalle, R.; et al. Large-Scale Differences in Microbial Biodiversity Discovery between 16s Amplicon and Shotgun Sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R.; et al. A Comparison of Sequencing Platforms and Bioinformatics Pipelines for Compositional Analysis of the Gut Microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A Review of Methods and Databases for Metagenomic Classification and Assembly. Brief. Bioinform. 2018, 20, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Fan, X.; Ying, J.; Chen, S. Emerging Trends and Research Foci in Gastrointestinal Microbiome. J. Transl. Med. 2019, 17, 67. [Google Scholar] [CrossRef]
- Antony-Babu, S.; Stien, D.; Eparvier, V.; Parrot, D.; Tomasi, S.; Suzuki, M.T. Multiple Streptomyces Species with Distinct Secondary Metabolomes have Identical 16S rRNA Gene Sequences. Sci. Rep. 2017, 7, 11089. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4fun: Predicting Functional Profiles From Metagenomic 16S rRNA Data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greay, T.T.L.; Gofton, A.W.; Zahedi, A.; Paparini, A.; Joll, C.A.; Ryan, U.M.; Linge, K.L.; Joll, C.A.; Ryan, U.M. Evaluation of 16S next-Generation Sequencing of Hypervariable Region 4 in Wastewater Samples: An Unsuitable Approach for Bacterial Enteric Pathogen Identification. Sci. Total Environ. 2019, 670, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Schloss, P.D. The Impact of DNA Polymerase and Number of Rounds of Amplification in PCR on 16S rRNA Gene Sequence Data. mSphere 2019, 4, e00163-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Updating the 97% Identity Threshold for 16S Ribosomal RNA OTUs. Bioinformatics 2018, 34, 2371–2375. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual Biology Across a Group Comprising More Than 15% Of Domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, M.M.; Botts, S.R.; Paschos, A.; Schellhorn, H.E. Temporal and Spatial Changes in Bacterial Diversity in Mixed Use Watersheds of the Great Lakes Region. J. Great Lakes Res. 2019, 45, 109–118. [Google Scholar] [CrossRef]
- Bag, S.; Saha, B.; Mehta, O.; Anbumani, D.; Kumar, N.; Dayal, M.; Pant, A.; Kumar, P.; Saxena, S.; Allin, K.H.; et al. An Improved Method for High Quality Metagenomics Dna Extraction From Human and Environmental Samples. Sci. Rep. 2016, 6, 26775. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A. FASTQC: Quality Control of Sequencing Data (Version 0.1.2). Available online: https://cran.r-project.org/web/packages/fastqcr/index.html (accessed on 24 November 2020).
- Gajer, P.; He, X.; Mccomb, E.; Gravitt, P.E.; Ghanem, K.G.; Brotman, R.M. Ultrahigh-Throughput Multiplexing and Sequencing of >500- Base-Pair Amplicon Regions on the Illumina HiSeq 2500 Platform. Appl. Environ. Sci. 2021, 4, e00029-19. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using Diamond. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. Megan Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Herbold, C.W.; Pelikan, C.; Kuzyk, O.; Hausmann, B.; Angel, R.; Berry, D.; Loy, A. A Flexible and Economical Barcoding Approach for Highly Multiplexed Amplicon Sequencing of Diverse Target Genes. Front. Microbiol. 2015, 6, 731. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the Miseq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference From Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R Package for Rarefaction and Extrapolation of Species Diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Soc. Am. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). Wiley StatsRef Stat. Ref. Online 2017, 557–574. [Google Scholar] [CrossRef]
- Jakaitiene, A.; Sangiovanni, M.; Guarracino, M.R.; Pardalos, P.M. Multidimensional Scaling for Genomic Data. In Advances in Stochastic and Deterministic Global Optimization; Springer International Publishing: Cham, Switzerland, 2016; Volume 107, pp. 229–242. ISBN 978-3-319-29973-0. [Google Scholar]
- Tzeng, J.; Lu, H.; Li, W.H. Multidimensional Scaling for Large Genomic Data Sets. BMC Bioinform. 2008, 9, 179. [Google Scholar] [CrossRef]
- Zhu, X.; Campanaro, S.; Treu, L.; Seshadri, R.; Ivanova, N.; Kougias, P.G.; Kyrpides, N.; Angelidaki, I. Metabolic Dependencies Govern Microbial Syntrophies During Methanogenesis in an Anaerobic Digestion Ecosystem. Microbiome 2020, 8, 22. [Google Scholar] [CrossRef]
- Roguet, A.; Newton, R.J.; Eren, A.M.; McLellan, S.L. Guts of the Urban Ecosystem: Microbial Ecology of Sewer Infrastructure. mSystems 2022, 7, e00118-22. [Google Scholar] [CrossRef] [PubMed]
- Batstone, D.J.; Puyol, D.; Flores-Alsina, X.; Rodríguez, J. Mathematical Modelling of Anaerobic Digestion Processes: Applications and Future Needs. Rev. Environ. Sci. Biotechnol. 2015, 14, 595–613. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, M.; Ma, X.; Gao, Q.; Wang, T.; Shi, X.; Zhou, J.; Zuo, J.; Yang, Y. High Variations of Methanogenic Microorganisms Drive Full-Scale Anaerobic Digestion Process. Environ. Int. 2019, 126, 543–551. [Google Scholar] [CrossRef]
- Yin, Q.; Zhu, X.; Zhan, G.; Bo, T.; Yang, Y.; Tao, Y.; He, X.; Li, D.; Yan, Z. Enhanced Methane Production in an Anaerobic Digestion and Microbial Electrolysis Cell Coupled System with Co-Cultivation of Geobacter and Methanosarcina. J. Environ. Sci. 2016, 42, 210–214. [Google Scholar] [CrossRef]
- Zhang, S.; Chang, J.; Liu, W.; Pan, Y.; Cui, K.; Chen, X.; Liang, P.; Zhang, X.; Wu, Q.; Qiu, Y.; et al. A Novel Bioaugmentation Strategy to Accelerate Methanogenesis Via Adding Geobacter Sulfurreducens PCA in Anaerobic Digestion System. Sci. Total Environ. 2018, 642, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.V. Identification of Sulfate-Reducing Bacteria Strains of Human Large Intestine. Stud. Biol. 2013, 7, 115–132. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, L.; Zhang, Z.; Chen, G.-H.; Jiang, F. Realizing High-Rate Sulfur Reduction Under Sulfate-Rich Conditions in a Biological Sulfide Production System to Treat Metal-Laden Wastewater Deficient in Organic Matter. Water Res. 2017, 131, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Roh, D.H. Phenylobacterium Muchangponense sp. nov., Isolated from Beach Soil, and Emended Description of the Genus Phenylobacterium. Int. J. Syst. Evol. Microbiol. 2012, 62, 977–983. [Google Scholar] [CrossRef]
- Verastigui, Y.; Cheng, J.; Engel, K.; Kolczynski, D.; Mortimer, S.; Lavigne, J.; Montalibet, J.; Romantsov, T.; Hall, M.; McConkey, B.; et al. Multisubstrate Isotope Labeling and Metagenomic Analysis of Active. mBio 2021, 5, e01157-14. [Google Scholar] [CrossRef]
- Puentes-Téllez, P.E.; Salles, J.F. Dynamics of Abundant and Rare Bacteria During Degradation of Lignocellulose from Sugarcane Biomass. Microb. Ecol. 2020, 79, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, M.Z.; Sok, W.; Choi, G.; Kim, S.Y.; Wee, J.H.; Im, W.T. Simplicispira hankyongi sp. nov., a Novel Denitrifying Bacterium Isolated Grom Sludge. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2020, 113, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Singh, R.; Eltis, L.D.; Mohn, W.W. Bacterial Contributions to Delignification and Lignocellulose Degradation in Forest Soils with Metagenomic and Quantitative Stable Isotope Probing. ISME J. 2019, 13, 413–429. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the Microbiome: Advantages of Whole Genome Shotgun Versus 16S Amplicon Sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmann, B.; Al-ghalith, G.A.; Shields-cutler, R.R.; Zhu, Q.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS A J. Integr. Biol. 2018, 22, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Wang, B.; Bai, X.; Luan, Y.; Fan, Y.; Michail, S.; Sun, F. 16S rRNA and Metagenomic Shotgun Sequencing Data Revealed Consistent Patterns of Gut Microbiome Signature in Pediatric Ulcerative Colitis. Sci. Rep. 2022, 12, 6421. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Vries, J.; Saleem, F.; Li, E.; Chan, A.W.Y.; Naphtali, J.; Naphtali, P.; Paschos, A.; Schellhorn, H.E. Comparative Analysis of Metagenomic (Amplicon and Shotgun) DNA Sequencing to Characterize Microbial Communities in Household On-Site Wastewater Treatment Systems. Water 2023, 15, 271. https://doi.org/10.3390/w15020271
de Vries J, Saleem F, Li E, Chan AWY, Naphtali J, Naphtali P, Paschos A, Schellhorn HE. Comparative Analysis of Metagenomic (Amplicon and Shotgun) DNA Sequencing to Characterize Microbial Communities in Household On-Site Wastewater Treatment Systems. Water. 2023; 15(2):271. https://doi.org/10.3390/w15020271
Chicago/Turabian Stylede Vries, Jacob, Faizan Saleem, Enze Li, Alexander Wing Yip Chan, James Naphtali, Paul Naphtali, Athanasios Paschos, and Herb E. Schellhorn. 2023. "Comparative Analysis of Metagenomic (Amplicon and Shotgun) DNA Sequencing to Characterize Microbial Communities in Household On-Site Wastewater Treatment Systems" Water 15, no. 2: 271. https://doi.org/10.3390/w15020271