

Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study
by
 , , and
, , and
Munazzah Tasleem
1,†,‡,
Wesam M. Hussein
2,
Abdel-Aziz A. A. El-Sayed
3 and
Abdulwahed Alrehaily
3,*,† 1
School of Electronic Science and Engineering, University of Electronic Science and Technology of China, Chengdu 610054, China
2
Chemistry Department, Faculty of Science, Islamic University of Madinah, Madinah 42351, Saudi Arabia
3
Biology Department, Faculty of Science, Islamic University of Madinah, Madinah 42351, Saudi Arabia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
‡
Current address: BIAltesse LLC, 5109 Silverton Ln, Louisville, KY 40241, USA.
Water 2023, 15(6), 1142; https://doi.org/10.3390/w15061142
Submission received: 20 February 2023
/
Revised: 11 March 2023
/
Accepted: 12 March 2023
/
Published: 15 March 2023
(This article belongs to the Topic Application of Smart Technologies in Water Resources Management)
Abstract
:In Saudi Arabia, seawater desalination is the primary source of acquiring freshwater, and groundwater contains a high concentration of toxic heavy metals. Chromium (Cr) is one of the heavy metals that is widely distributed in the environment, particularly in the groundwater of Madinah. Diverse techniques are employed to eliminate the toxicity of heavy metals from the environment, but, lately, the focus has shifted to biological remediation systems, due to their higher removal efficiencies, lower costs, and more ecologically benign characteristics than the conventional methods. Providencia bacteria engage in a variety of adsorption processes to interact with heavy metals. In this study, we aim to investigate the role of potential active site residues in the bioengineering of chromate reductase (ChrR) from Providencia alcalifaciens to reduce the Cr to a lesser toxic form by employing robust computational approaches. This study highlights Cr bioremediation by providing high-quality homology-modeled structures of wild type and mutants and key residues of ChrR for bioengineering to reduce the Cr toxicity in the environment. Glu79 is found to be a key residue for Cr binding. The mutant models of Arg82Cys, Gln126Trp, and Glu144Trp are observed to establish more metallic interactions within the binding pocket of ChrR. In addition, the wild type ChrR (P. alcalifaciens) has been found to be unstable. However, the mutations stabilized the structure by preserving the metallic contacts between the critical amino acid residues of the identified motifs and the Cr(VI). Therefore, the mutants discovered in the study can be taken into account for protein engineering to create reliable and effective enzymes to convert Cr(VI) into a lesser toxic form.
1. Introduction
Water contamination poses a significant risk to all living things, including mankind, terrestrial animals, aquatic animals, and plants. The importance of having access to clean water, proper sanitation, and the wise management of freshwater ecosystems is being recognized by an increasing number of academics and scientists. This is important for several reasons, including human health, the long-term viability of the environment, and economic growth [1]. The vast majority of the population in KSA is dependent on non-renewable resources, as the desalination of seawater in that country is sufficient to meet the needs of the people who reside in and around the coastal areas [2].
The concentration of toxic metal ions in the groundwater of KSA has been extensively studied, revealing high concentrations of aluminum (Al), arsenic (As), barium (Ba), cadmium (Cd), cobalt (Co), chromium (Cr), copper (Cu), iron (Fe), mercury (Hg) [3], lithium (Li), manganese (Mn), nickel (Ni), lead (Pb), selenium (Se), and zinc (Zn) [4].
Different regions in Madinah were examined for the presence of Cr as one of the toxic heavy metals in the groundwater. According to WHO [5], the concentration of Cr in Wadi Al Aqiq is 1–146 g/L [6], and Uhud, Quba, Al Aqool, and the neighborhood surrounding the Prophet’s Holy Mosque were shown to be high (0.011–0.11 mg/L) [7,8]. Chromium exists in seven distinct oxidation states, from 0 to VI, and three forms, namely, Cr(0), Cr(III), and Cr(VI), which are found in high concentrations in nature [9,10,11,12]. The chromium oxidation states determine its toxicity, mutagenesis potential, and other biological impacts. As a result of its oxidizing nature, hexavalent chromium Cr(VI) is more dangerous, carcinogenic, teratogenic, mutagenic, and mobile than trivalent chromium Cr(III) [12,13]. Cr is categorized as a human carcinogen by the International Agency for Research on Cancer (IARC) due to its potential for mutagenesis effects (Group 1 category). The primary route of chromate ingestion is through the consumption of drinking water [14]. Conventional physicochemical techniques to remove chromium from contaminated environments, such as wastewater, include ion exchange, chemical precipitation, floatation, flocculation, electrolysis, coagulation, adsorption, membrane filtration, reverse osmosis, and photocatalysis [15,16,17]. The attention has switched to biological remediation systems with relatively high removal efficiencies, low cost, and ecologically benign properties since the removal of chromium by conventional methods is ineffective. Utilizing the capacity of microorganisms or plants to bind or change heavy metals, biotransformation and biosorption are frequently applied technologies [15,18].
In order to detoxify and remove Cr-pollutants under a variety of experimental situations, bioremediation, which makes use of local microorganisms, is a straightforward, affordable, and environmentally beneficial approach [19]. The majority of bioremediation techniques, such as biosorption, remove hazardous heavy metal ions with little to no secondary waste [20].
A genus of rod-shaped, Gram-negative bacteria called Providencia is a member of the Enterobacteriaceae family. The strong adsorption of lead, aluminum, cobalt, copper, and chromium by a family of Providencia bacteria has been demonstrated [21,22,23,24]. It is noteworthy that Providencia bacteria interact with these metals through a variety of adsorption techniques. For instance, Providencia vermicola KX926492 does not significantly exhibit Pb(II) adsorption on cell surfaces and, instead, largely accumulates Pb(II) in the form of lead sulfite through periplasmic sequestration within cells [21]. While Providencia alcalifaciens 2 EA absorbed Pb(II), phosphatase catalyzed the formation of insoluble lead orthophosphate of Pb9(PO4)6 on cell surfaces [22]. It has proven possible to isolate P. alcalifaciens from a range of living species, particularly cows, dogs, and chickens, as well as from soil, water, and sewage [25].
In this study, in silico studies are preferred over conventional studies, which are expensive, labor-intensive, and time-consuming. Chromate reductase (ChrR) from P. alcalifaciens was analyzed to establish the existence of super-secondary structures, family, and phylogenetic relationships. Homology models built from structurally similar homologs would be the optimal starting point to comprehend the binding interaction with Cr(VI) in the absence of the three-dimensional structure of ChrR from P. alcalifaciens. To create a stable and effective enzyme for bioremediation, the most efficient residues for protein engineering were determined by analyzing the mutant models. The research describes ligand binding and the structural properties of the ligands that regulate the biological activities of the bacteria, which will support the bioremediation of Cr by P. alcalifaciens bioremediation. Structural and dynamic studies will contribute to building stronger chromate reductase-related enzymes to reduce environmental chromate pollution.
2. Materials and Methods
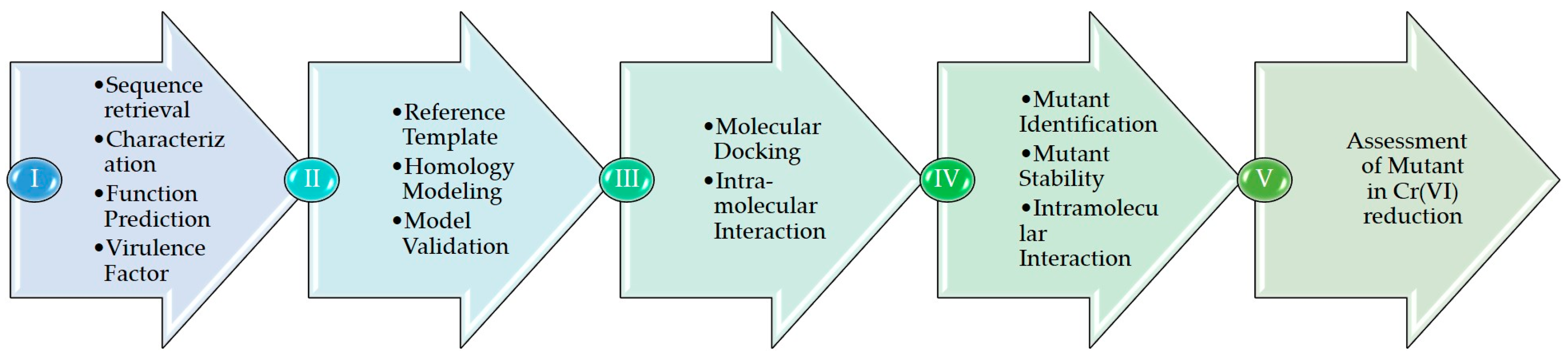
Figure 1 depicts the in silico approach adopted for annotating the sequence. Step I mention the sequence, its characterization, function, virulence factor, and its comparison with other species. The second step involves identifying reference templates, and the final homology models are assessed using a variety of bioinformatics tools. Step III includes docking and studying the intramolecular interactions between ChrR and Cr(VI). Step IV aims to predict mutant residues, mutant model generation, and docking with Cr(VI). Step V comprises an assessment of the interactions between Cr(VI) and wild type and mutant models.
2.1. ChrR Sequence Annotation
Using the online data retrieval system from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/ (accessed on 10 December 2022)), ChrR protein sequences from P. alcalifaciens were obtained in the FASTA format [26]. The precise assignment of protein roles was accomplished using various bioinformatics approaches. To measure the physical and chemical properties of ChrR, the Expasy ProtParam (https://web.expasy.org/protparam/ (accessed on 10 December 2022)) service was used. The virulent protein sequence was predicted using the potent algorithms of VirulentPred (http://203.92.44.117/virulent/ (accessed on 10 December 2022)), MP3 (http://metagenomics.iiserb.ac.in/mp3/index.php (accessed on 10 December 2022)), and VICMPred (https://webs.iiitd.edu.in/raghava/vicmpred/ (accessed on 10 December 2022)). Support vector machine (SVM) methods are used by the VirulentPred program to precisely predict the virulence factors of bacteria [27]. The function of proteins found in gram-negative bacteria can be predicted by using the web server VICMpred [28]. The MP3 program is able to identify potentially dangerous proteins in metagenomic datasets [29]. PSI-BLAST (https://blast.ncbi.nlm.nih.gov/ (accessed on 10 December 2022)) [22] was used to identify structurally related proteins in the Protein Data Bank (PDB) database (https://www.rcsb.org (accessed on 10 December 2022)) [23].
The retrieved protein sequence was used to identify the ortholog sequences, family, and domains, and their functions were subsequently evaluated. The eggNOG 5.0 (http://eggnog.embl.de/ (accessed on 11 December 2022)) was utilized to identify the orthologs of ChrR in 4445 bacterial species [30].
InterProscan (https://www.ebi.ac.uk/interpro/about/interproscan/ (accessed on 11 December 2022)) and HMMER (http://hmmer.org/ (accessed on 11 December 2022)) use the hmmscan program to identify the domains in a sequence. Protein family databases frequently use profile HMM models. The significance of a hit is depicted by an individual E-value as though it were the lone domain or hit found. The conditional E-value aims to quantify the statistical significance of each domain after confirming that the target sequence is a true homolog. PANTHER, ProDom, Pfam, PRINTS, Prosite, SUPERFAMILY, and other highly developed protein signature recognition algorithms are included in InterProscan.
The Pfam (http://pfam-legacy.xfam.org/ (accessed on 11 December 2022)) database [24] and CATH (http://cathdb.info/ (accessed on 12 December 2022)), the Protein Structure Classification Database at UCL [25], were used to classify the protein sequence families [31]. For predicting the domains, the HMMER web server online tools were used. The HMMER server is based on profiles from hidden Markov model (HMM) libraries and provides rapid searches against well-known sequence databases. PANNZER (Protein ANNotation with Z-scoRE) (http://ekhidna2.biocenter.helsinki.fi/sanspanz/ (accessed on 12 December 2022)) performed the function prediction. PANNZER is a completely automated service, which annotates prokaryotic and eukaryotic proteins of undetermined function. The program is intended to determine a functional description (DE) and GO classes, along with DeepGoWEb (https://deepgo.cbrc.kaust.edu.sa/deepgo/ (accessed on 12 December 2022)). Protein families are considered to be distinguished by specific protein sequence motifs, which makes it simpler to speculate on the potential functions of the proteins. Motifs are crucial to enzyme function because of their association with catalytic reactions. The Mutiple EM for Motif Elicitation (MEME v.5.5) tool (https://meme-suite.org/meme/ (accessed on 12 December 2022)) was also used for the motif searching [32].
Multiple sequence alignment (MSA), Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 13 December 2022)), and Phylogeny.fr (https://www.phylogeny.fr/ (accessed on 13 December 2022)) were utilized to locate the conserved regions throughout the sequence. Phylogeny.fr is an unrestricted and user-friendly web service for regenerating and evaluating evolutionary relationships among molecular sequences. It executes and connects multiple bioinformatics programs in order to create a comprehensive phylogenetic tree from a group of sequences. The advanced mode pipeline was applied, and the settings of Phylogeny.fr were used in which MUSCLE was used to generate multiple sequence alignments; alignment curation was performed by Gblock without allowing many contiguous non-conserved positions; and PhyML (http://www.atgc-montpellier.fr/phyml/ (accessed on 13 December 2022)) was used to construct a phylogenetic tree employing 100 bootstrap replicates and the Jones–Taylor–Thornton matrix substitution model.
Sequence-based NetSurfP-2.0 (https://services.healthtech.dtu.dk/services/NetSurfP-2.0/ (accessed on 14 December 2022)) employs a convolutional and long short-term memory neural network architecture that was developed using protein structural solutions as training data. Using a single integrated model, NetSurfP-2.0 predicts the secondary structure, solvent accessibility, structural disorder, and backbone dihedral angles for each residue in the input sequences. The most recent version of the well-known JPred protein secondary structure prediction program, JPred4 (http://www.compbio.dundee.ac.uk/jpred4 (accessed on 14 December 2022)), provides predictions using the JNet algorithm, one of the most accurate methods for predicting secondary structures.
2.2. Homology Model and Validation of ChrR
A tertiary structure for the query sequence was generated through in silico approaches with experimentally comparable accuracy to establish the structure–function association. By employing a reference template whose tertiary structure has been experimentally validated, the structures were built using the homology modeling technique. Structures are more likely to be conserved across time than sequences are, and sequences that are similar tend to adopt similar structures. As a result, generated 3D structures will serve as a foundation for understanding the unique functions of the unannotated protein sequences and will shed considerable light on interactions.
In order to retrieve the template, a sequence similarity search against the PDB database was conducted using the PSI-BLAST tool, which produced a list of homologous sequences to the query sequence. The sequence with the highest sequence identity, the most extensive query coverage, and the lowest E-value score was identified as the reference template. The coordinates of the template structure were acquired from the Protein Data Bank based on the BLAST [33].
The following five homology modeling tools were chosen to create a three-dimensional structure: (i) GENO3D (geno3d-prabi.ibcp.fr (15 December 2022)) (https://geno3d-prabi.ibcp.fr/cgi-bin/geno3d_automat.pl?page=/GENO3D/geno3d_home.html (accessed on 15 December 2022)) is a technology for modeling comparative protein structures while satisfying spatial constraints (dihedral and distance) [34]; (ii) IntFOLD (https://www.reading.ac.uk/bioinf/IntFOLD/ (accessed on 15 December 2022)) provides built-in estimations of model accuracy (EMA), protein structural domain borders, naturally unstructured or disordered areas in proteins, and protein–ligand interactions to predict protein tertiary structures [35]; (iii) Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/ (accessed on 16 December 2022)) is an online homology detection program that generates the structure and predicts binding sites in the structure [36]; (iv) Robetta is a protein structure prediction tool that is continuously assessed by CAMEO [37]; (v) I-TASSER (Iterative Threading ASSembly Refinement) (https://zhanggroup.org/I-TASSER/ (accessed on 16 December 2022)) is a hierarchical technique for predicting protein structure and annotating structure-based functions [38].
Two competent tools were used to assess the quality of the models: the ProQ Protein Quality Predictor (https://proq.bioinfo.se/ProQ (accessed on 20 December 2022)), a neural network-based method for calculating the LGscore and MaxSub score [39]; and the Protein Structure Validation Suite (PSVS) (https://montelionelab.chem.rpi.edu/PSVS/psvs2/ (accessed on 20 December 2022)). The PDB validation software, Verify3D Prosa II, RPF, PROCHECK, MolProbity, and other structure-validation tools are the various structure quality evaluation tools that the PSVS incorporates. The PSVS provides conventional constraint studies, PDB validation good-ness-of-fit statistics between the structures and experimental data, and knowledge-based structure quality scores for database integration. Global quality measurements are displayed as Z scores, based on a calibration with a set of high-resolution X-ray crystal structures. Using the molecular graphics technology of Discovery Studio Visualizer and PyMol Molecular Graphics System Version 2.5, a comprehensive structural analysis was carried out on the validated optimal structure.
2.3. Cr(VI) Molecular Docking and Intramolecular Interaction Studies
Taking into account the high sequence identity and structural similarity between the template (PDB ID: 1RTT_A) and the target protein, the active site was identified. To facilitate the identification of the binding site, the active site residues were investigated using MSA [40,41]. As a result, the optimum site for ligand docking was selected to be the projected binding site. Using the CDOCKER program from DS, docking studies were conducted to determine the optimal binding mechanism of the Cr(VI) to the ChrR protein [42]. Hydrogen was added, followed by energy minimization, charge correction, and side-chain refinement, to complete the preparation of the ligand and the protein. The studies reveal that the highly attached FMN molecule is near the metal-binding region in the Gh-ChrR [41]. The active site was uploaded to define the active site, and its x, y, and z coordinates are 17.015425, −0.051868, and 66.484199, respectively. It also has a radius of 16.242105 Å. The close intramolecular interactions between Cr(VI) and ChrR that took place between 2.5 and 3.5 Å were evaluated using the View Interaction module from DS to ascertain the stability of the docked complex. The Calculate Binding energy tool was applied to estimate the binding energy of the receptor and the ligand.
2.4. Generation and Molecular Docking of In Silico Mutant of ChrR to Analyze Intramolecular Interaction with Cr(VI)
Several investigations revealed site-directed mutations in E. coli ChrR that enhanced the activity of chromate reductase [43]. By changing the residues that give a protein stability to active site residues, mutants were constructed. By applying the “Mutagenesis” module of PyMol Molecular Graphics System Version 2.5, mutated models were constructed, and the side chains were optimized, followed by energy minimization and an evaluation of stability. To evaluate binding affinity and intramolecular interactions, the most stable structure was docked with Cr(VI).
3. Results
3.1. ChrR Sequence Annotation
The amino acid sequence of the Providencia alcalifaciens chromate reductase containing 185 amino acid residues was obtained from the UniprotKB database (https://www.uniprot.org (accessed on 10 December 2022)), using the accession number A0A4R3NKM6. ChrR from P. alcalifaciens, as determined by a Uniprot analysis, contains an FMN reductase domain from positions 6 to 146. The estimated physicochemical properties of the query sequences are displayed in Table 1. A0A4R3NKM6 has an estimated instability index of 41.23. By using VirulentPred at a 0.5 threshold, Predict Pathogenic Proteins in Metagenomic Data (MP3), and VICMpred prediction approaches, the virulence of the ChrR was predicted. As demonstrated in Table 2, the consensus analyses of the sequence confirmed it to be a non-pathogenic protein.
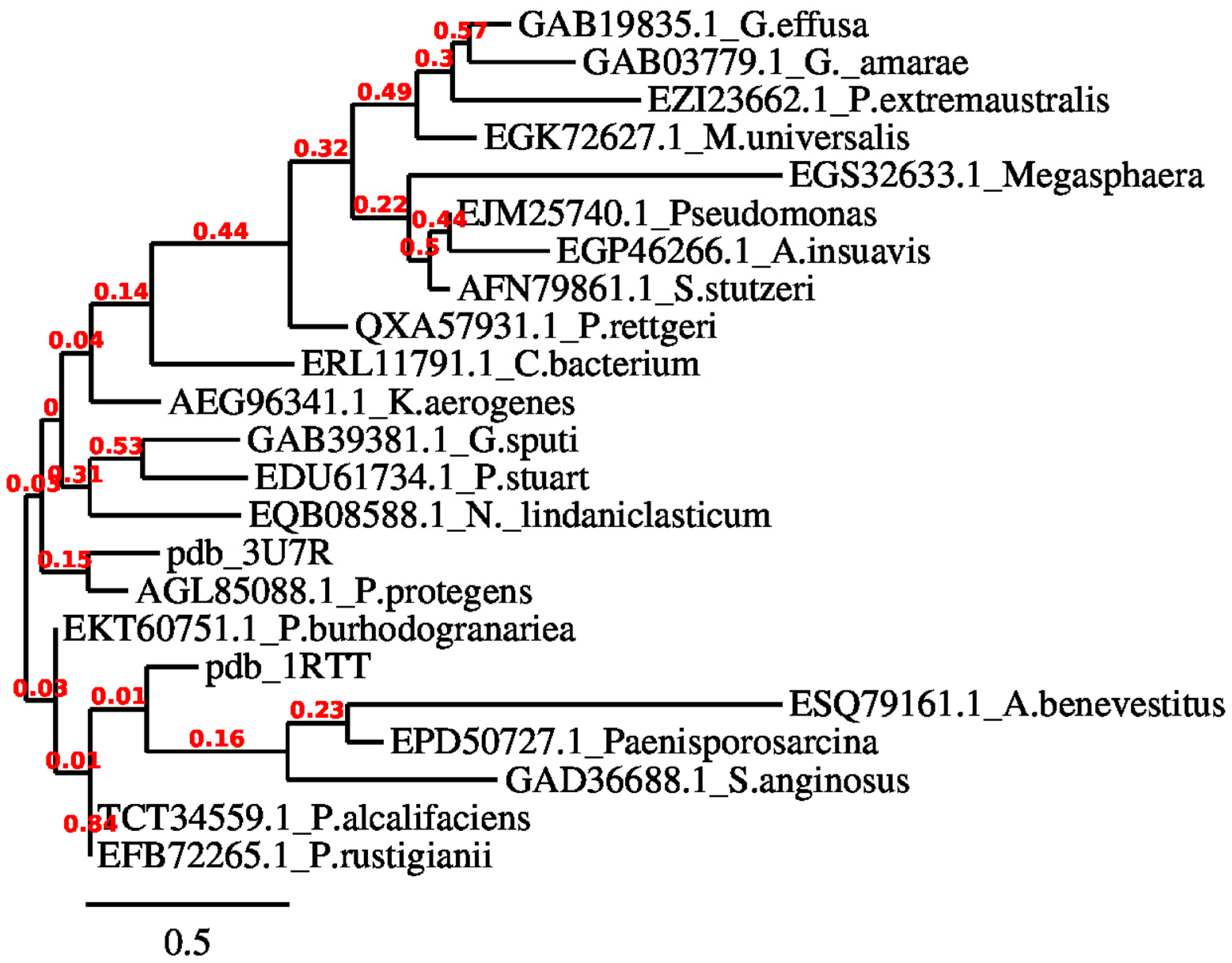
Various biological research domains, such as comparative genomics, functional prediction, lateral gene transfer detection, or the discovery of novel microbes, depend on reconstructing the evolutionary history of nucleotide sequences through phylogenetic analysis. In this study, the eggNOG 5.0 program was employed to determine the orthologous chromate reductase sequences in distinct bacterial species. As illustrated in Figure 2, orthologs for ChrR were identified, enabling the phylogenetic analysis and multiple sequence alignment to identify the conserved sites and emphasize the evolutionary relationship. To determine the ancestral states, the approximate likelihood ratio test (aLRT) and Jones–Taylor–Thornton matrix-based model were applied. A variety of likely amino acids (states) are shown for each ancestral node on the basis of their approximation of likelihood at site 1. For each node, just the most probable state is shown. The initial tree(s) for the heuristic search were constructed using the JTT model’s pairwise distances and maximum likelihood, and the topology with the highest log likelihood score was identified. As seen in Figure 3, the orthologous amino acid sequences were chosen for research.
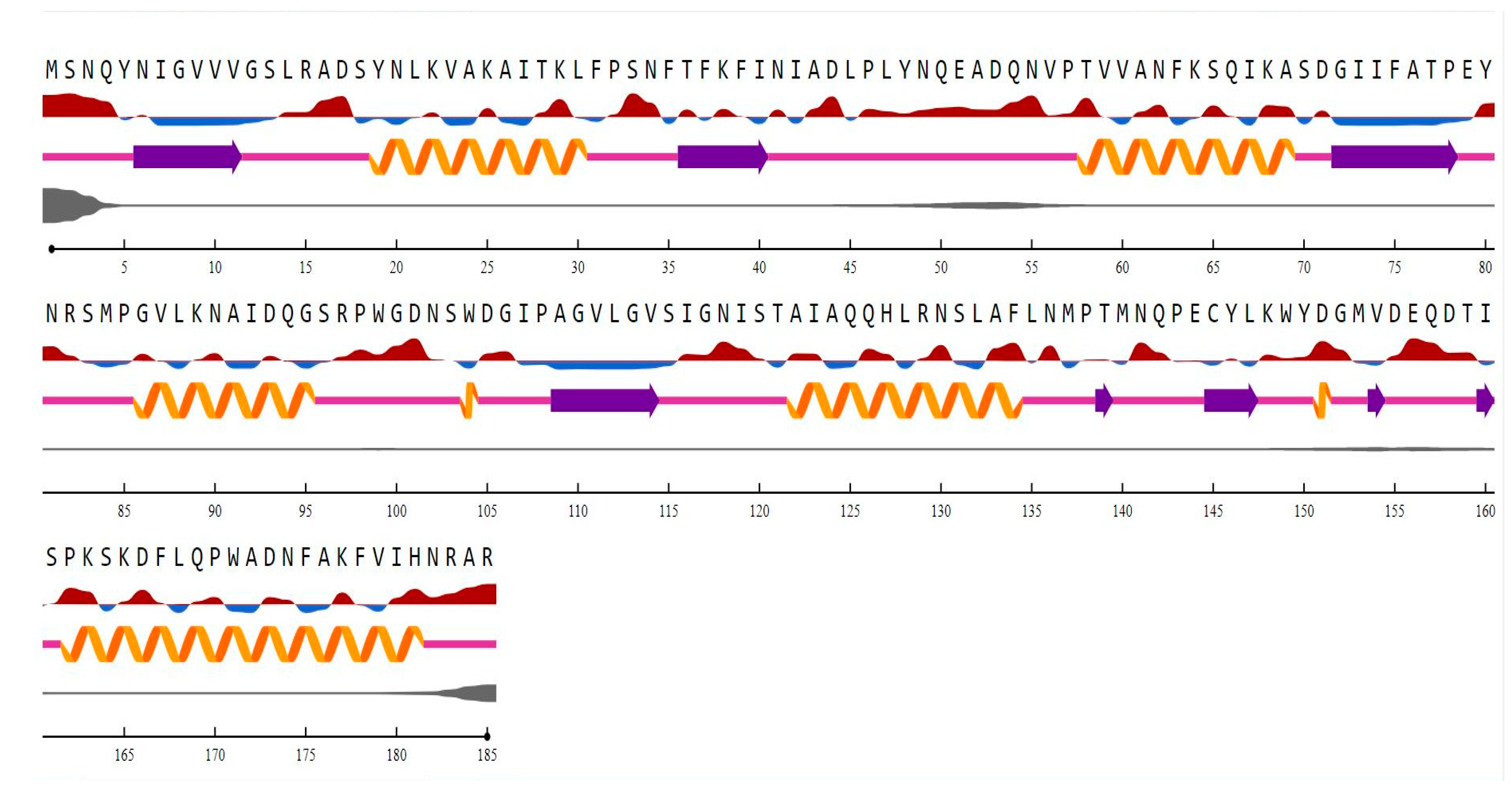
The motifs identified by using MEME v5.5 are: “FATPEY” (75 to 80), “WGDNSWDG” (99 to 106), and “HLRNSLAFLNMPT” (127 to 139). The domain “NADPH-dependent FMN reductase” was found in regions 1 to 156. The InterProsan revealed it to belong to the flavoprotein superfamily and the “FMN-red” family from 6 to 146 [1]. When the structure of a novel protein is unknown, a reliable approach for predicting its secondary (and eventually tertiary) structure is required. There are various computer methods for predicting the secondary structure of a given protein sequence, and they all aim to distinguish between the helix (H), strand (S), and coil/loop (C) regions. The secondary structure content and the disordered regions in the ChrR in P. alcalifaciens, shown in Figure 4, describe a higher content of helices than sheets.
3.2. Homology Model and Validation of ChrR
The A0A4R3NKM6 protein sequence was found to be most similar to the ChrR from Paracoccus denitrificans (PDB ID: 3U7R_A), a NADPH-dependent FMN reductase, with a similarity of 40.00% and a maximum query coverage of 94%. The template sequence for homology modeling of ChrR P. alcalifaciens was selected as the putative NADH-dependent reductase from Pseudomonas aeruginosa, with the second highest similarity of 39.75% and a sequence coverage of 86%. Phyre2, Geno3D, Robetta, IntFold, and I-Tasser were used to create five structures of ChrR from P. alcalifaciens. Since no single quality parameter is ideal, we employed two distinct quality evaluation tools to confirm the protein structure’s quality. ProQ Protein Quality Predictor and PSVS were applied to validate the best model generated by each tool. The outcomes of various procedures used for verification consistently demonstrated the excellent quality of the suggested models. The correct models were assessed based on the LGscore of >1.5 and MaxSub of >0.1 [39], where the most “significant” non-continuous portion of a model is determined via LGscore, and the structural p-values are used to calculate the similarity [44]. MaxSub generates a single normalized score that reflects the model’s quality by determining the greatest subset of Cα atoms in a model that superimposes “well” over the experimental structure [45]. The Phyre2 model showed the highest LGscore, while all the other modeled structures showed LGscores of >7. However, the MaxSub of all the models was <0.1. The Ramachandran plot study, where a good model is predicted to have over 90% of residues in the most favored region, indicated that the IntFold homology model was of the highest quality; however, the Phyre2 and Robetta models scored close to 90% in the most favored region. The Phyre2, Geno3D, and Robetta models were found to possess the lowest RMSD. The Verify3D tool evaluates the likelihood that the amino acid sequence has the observed three-dimensional packing [46]. The PROSAII tool computes the pseudo-energy of the pair-wise interactions based on the spatial spacing of residues [47]. The MolProbity tool calculates and depicts undesirable interactions, atomic overlaps, and variations in the Cβ position [48]. The Verify3D, PROSAII, and MolProbity scores indicate the models to be of good quality. All of the protein quality evaluation tools strongly suggest the Phyre2-generated model was the best quality, as shown in Table 3.
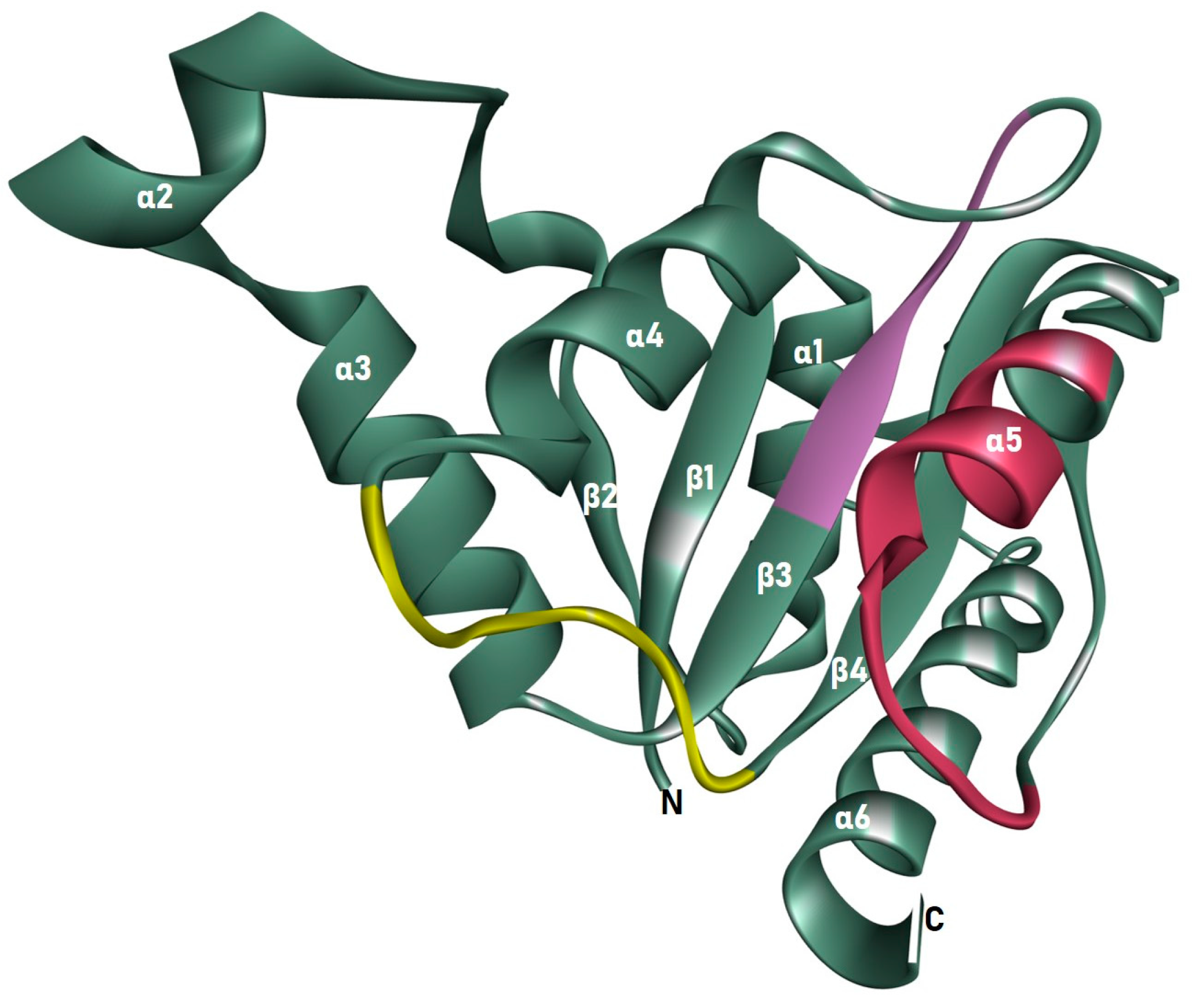
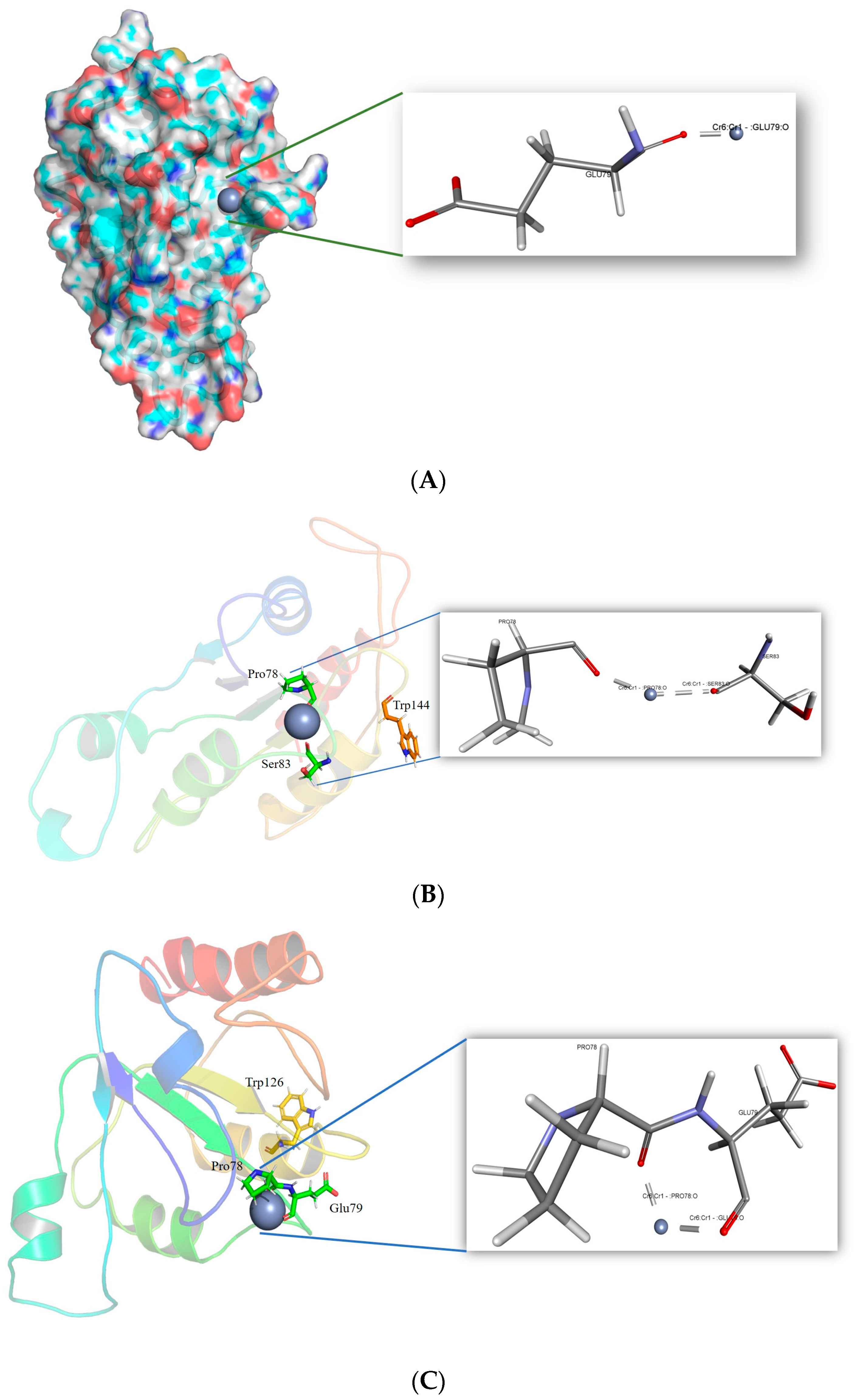
The 178 residues from Tyr5 to Asn182 make up the homology-modeled structure. The model was composed of six helices, four sheets, and seven loops. It was discovered that the structure contains an NADPH-dependent FMN reductase domain. It is a member of the superfamily of flavoproteins. All four sheets were found to be organized parallel to one another in the center of the structure, surrounded on one side by the 2, 3, and 4 helices, and on the other side by the 1 and 5. The FMN binding site is located in the cavity created by loops 1, 2, and 3, close to the c-terminus end of the sheet. As illustrated in Figure 4, three motifs were discovered, two in the secondary region, and one in the loop. Cr(VI) is localized in the binding pocket of the ChrR with a CDOCKER score of 29.89 and a CDOCKER Interaction Energy of 12.69 kcal/mol. Cr(VI) was shown to interact with the conserved Glu79 O atom at a distance of 1.52 Å, as illustrated in Figure 5.
3.3. Generation and Molecular Docking of In Silico Mutant of ChrR to Analyse Intramolecular Interaction with Cr(VI)
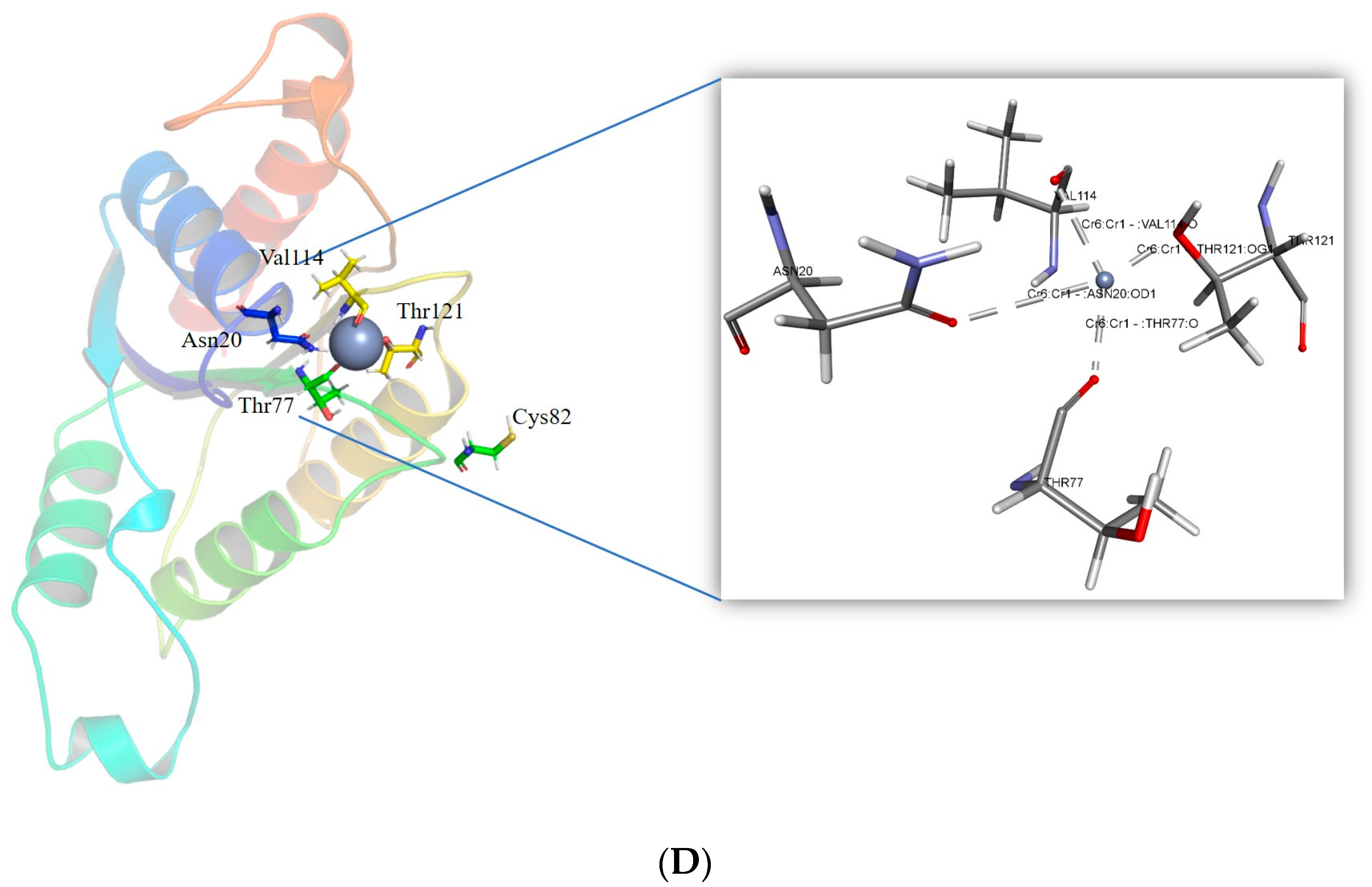
The corresponding residues to the reported mutant residues Tyr85, Arg125, Tyr128, and Glu146 were found using sequence alignment [30]. Arg82, Ile123, Gln126, and Glu144 are the corresponding residues. The most stable mutant of these residues was created. The II of the mutants was determined by the ProtParam tool. The estimated II of Arg82Cys is 38.90, Gln126Trp is 41.47, Ile123Leu is 37.76, and Glu144Trp is 37.76. The Cr(VI) was docked in the binding pocket of the mutant models to evaluate the interaction. Mutating Arg82 to Cys resulted in a stabilizing effect, with a mutation energy of −0.82 kcal/mol. Mutating Ile123 had a neutral and destabilizing effect. The stabilizing effect of the Gln126 to Trp and Tyr mutation was observed. However, the mutant Gln126Trp possesses the lowest mutation energy, −1.59 kcal/mol. As demonstrated in Table S1, the mutation of Glu144 to Trp had a stabilizing impact with mutation energy of −193 kcal/mol. The Arg82Cys, Gln126Trp, and Glu144Trp mutant models were produced using the “Build Mutant” procedure from DS. All the mutant structures were optimized for energy efficiency. Among the potential docking models for the Cr(VI), the one with the least amount of energy was selected. The mutant model Glu144Trp was observed to form a metallic interaction with the conserved residue Glu79. The mutant Arg82Cys was observed to form close metallic interactions with four residues: Asn20, Val114, Thr121, and a conserved residue located in the motif, “FATPEY”. As seen in Figure 6, all the mutants form a higher number of metallic interactions with the conserved residues.
4. Discussion
ChrR from P. alcalifaciens has an instability index calculated from its sequence, which is greater than 40, indicating that it is an unstable protein. However, it was discovered that the II of the mutant proteins Arg82Cys, Ile123Leu, and Glu144Trp was <40, indicating that the mutants constituted stable proteins. Consensus assessments of the enzyme sequence pathogenicity demonstrated that it was nonpathogenic and acceptable for human utilization. The MSA revealed that the amino acids associated with the motifs are largely conserved among the orthologs. This generated phylogenetic tree showed that ChrR from P. rustigiani was closely related to P. alcalifaciens. It is commonly accepted that the functional specialization of proteins is highly conserved among orthologs. By using this concept as a guide, the amino acids that regulate the selectivity of protein–ligand interactions were discovered. The research on molecular recognition mechanisms and the development of efficient proteins and drugs depend on identifying these residues [49]. In addition, ChrR from P. aeruginosa (PDB ID: 1RTT) shared FMN-dependent NADPH:quinone reductase and was found to be closely related. From regions 1 to 156, the domain “NADPH-dependent FMN reductase” was discovered. According to the InterProScan, it is a member of the 6–146 “FMN-red” family and the flavoprotein superfamily. The ChrR protein possesses a flavodoxin-like fold, which is composed of five parallel sheets joined by helices that surround the sheet. Flavodoxins are electron-transporting proteins that function in a number of electron transport systems. They are functionally comparable to ferredoxins since they bind one FMN molecule, which serves as a redox-active prosthetic group, and a sulfite reductase [NADPH] flavoprotein alpha-component, flavorubredoxin, FMN-dependent NADH-azo compound oxidoreductases, ribonucleotide reductase stimulatory proteins, and glutathione-regulated potassium-efflux system ancillary. FMN reductase uses NADPH or NADH to reduce flavins. It participates in a variety of biological processes that use facilitated flavin for particular purposes, such as bacterial bioluminescence. The flavodoxin is made up of structural domains that have a fold that is similar to that of flavodoxins and FMN-dependent NADH-azo compound oxidoreductases, which are present in many proteins. An open twisted alpha/beta structure made up of five parallel beta-sheets joined by encircling alpha helices characterizes the flavodoxin fold. Flavodoxins are proteins that function in multiple electron transport systems and convey electrons. In their capacity to bind one FMN molecule, which serves as a redox-active prosthetic group, they are equivalent to ferredoxins [50]. Therefore, the presence of the FMN binding domain ensures that the Cr will bind to the FMN and be reduced. The Rossman fold topology, one of the most common and widely distributed super-secondary structures, is present in the sequence. The structure is made up of an alpha-helical segment that alternates with segments of the hydrogen-bonded beta strands to form a beta-sheet. The initial beta-alpha-beta fold is the best-preserved part of the Rossmann folds. Due to the fact that it interacts with the ADP portion of dinucleotides such as FAD, NAD, and NADP, this area is sometimes referred to as an “ADP-binding fold.” [51]. Detailed computational research revealed the importance of the ChrR sequence in metal ion binding, iron-sulfur cluster binding, oxidoreductase activity, and catalytic activity.
Understanding a protein’s intricate function requires knowledge of the protein’s three-dimensional structure. In comparison to the three-dimensional structure prediction, the secondary structure prediction is typically accurate and much easier to solve [52]. The accuracy of methods to determine the protein’s three-state secondary structure, which includes an α-helix, a β-strand, and a coil, has grown over the past 30 years, going from about 50% in 1983 to over 80% today [53,54,55]. Secondary structure predictions offer significant constraints for fold-recognition methods, even if they are not as useful on their own as a complete three-dimensional model [55]. In addition to aiding in the discovery of functional domains, secondary structure predictions can guide the appropriate implementation of site-specific or deletion mutation studies. The secondary structure content computed by NetSurf (sheets 16.75% and helices 37.29%) indicates a compact structure.
It has been reported that ChrR catalyzed the quantitative transition of chromate to Cr(III). Since Cr(III) was the end product of the reactions, it is clear that the enzyme did more than just catalyze the one-electron reduction of Cr(VI) to Cr(V) [56]. Consequently, the molecular docking method evaluated Cr(VI) binding and intramolecular interactions. Organic compounds are the primary electron donors for reducing Cr(VI). It has been found that inorganic compounds can, nonetheless, function as reducing equivalents. Important electron donors for the reduction of Cr(VI) include fructose, lactose, lactate, glucose, pyruvate, citrate, acetate, glycerol, formate, reduced glutathione, NADH/NADPH, and other compounds. Gluconacetobacter hansenii (Gh)-ChrR, an FMN-containing chromate reductase, undergoes structural modifications as a consequence of the interaction of the chromate anion and NADH. In the absence of this rearrangement, the binding site would have been filled by the electron donor, preventing both species from attaching concurrently for efficient enzyme cycling. Additionally, it is extremely common for bacteria to reduce Cr(VI), utilizing glucose as the donor material. According to the present work, Cr(VI) can attach to the binding pocket and generate metal interactions with Glu79, a significant residue in the motif.
The ability to catalyze reactions, selectively bind ligands, and serve as materials and food additives, engineered proteins, and enzymes in particular, are finding increasing application in a variety of different business sectors. Along with the growth in the variety of contexts in which modified proteins might be useful, there has been an accompanying surge in the desire to design or create proteins with improved levels of stability, activity, and specificity. With the advancement in the application of protein technology, it will become vital to realize the potential benefits of regulating remote locations [57]. Eswaramoorthy, S. et al. reported mutations with increased chromate reductase activity compared to the wild type. In the present study, ChrR from P. alcalifaciens was compared to these mutants.
Bioremediation techniques such as biostimulation, which are part of bioremediation strategies, are examples of possible bioremediation technologies [58]. For the purpose of encouraging the growth of microorganisms, this technique involves adding nutrients to the surrounding environment, which may include aquifers. The modification enhances bioremediation, but an excessive buildup of biomass could potentially block subsurface pores, which would reduce the efficiency of the cleanup. Garbage that is mixed together makes it more difficult for bacteria and enzymes to do their job. Due to this, biostimulation of such locations is ineffective. The formation of toxic intermediates during chromate reduction makes chromate clean-up more difficult since these intermediates are toxic to the microorganisms used in the remediation process. Techniques involving genetic modification and protein engineering present a potential solution to these issues [59]. By employing specific promoters, the desired genes of slow-growing bacteria can be produced to their full potential, decreasing biomass accumulation and clogging. Protein engineering of bacterial ChrR can develop enzymes with enhanced chromate-reduction efficiency, reduced toxicity to the bacteria conducting the remediation, and the ability to function in the presence of additional contaminants [60].
The corresponding residues to the identified key residues that can enhance the functioning of the enzyme were determined by applying in silico approaches. The extensive analysis revealed the mutant that will lead to the stability of the protein. In addition, the mutant residues were found to form a higher number of metallic interactions with the residues in the binding pocket of the ChrR, indicating better binding with Cr(VI) to convert it to a less-toxic form Cr(III).
5. Conclusions
A variety of bioremediation strategies, in particular those based on microorganisms, have been developed as a result of the challenging problem of removing Cr(VI) contamination from the environment. An adaptable bacterium is P. alcalifaciens. This bacterium is evolving into a cell factory, producing organic compounds with a variety of biological activities, owing to its strong metabolism, resistance to toxins and oxidative stress, and adaptability to genetic manipulation. The system is established in the current study for better binding with Cr(VI) to reduce it to Cr(III), and a three-dimensional structure of the ChrR from P. alcalifaciens and the mutant models are provided. In fact, the information presented here indicates that Glu79 plays a vital function in binding with Cr(VI). We have demonstrated that Cr(VI) can still be reduced to Cr(III) using the mutant models with the amino acid substitutions Arg82Cys, Gln126Trp, and Glu144Trp. It was found that the P. alcalifaciens wild type ChrR was unstable. However, the mutations preserved the metallic contact formation between the key amino acid residue of the discovered motifs and stabilized the structure. These mutants can be further tested for protein engineering in order to introduce stable ChrR that can endure and lower the Cr(VI) in Madinah, Saudi Arabia’s harsh climate. Our research will concentrate on the involvement of other non-pathogenic microbes in the bioremediation of heavy metals contaminating the Madinah groundwater since our findings showed that ChrR from P. alcalifaciens is a potential enzyme for bioengineering and bioremediation of Cr.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/w15061142/s1, Table S1: Mutants stable residue and their energy.
Author Contributions
Conceptualization, A.A. and M.T.; methodology, M.T. and A.A.; software, M.T. and A.A.; validation, M.T. and A.A.; formal analysis, M.T. and A.A.; investigation, M.T. and A.A.; resources, M.T., A.-A.A.A.E.-S. and A.A.; writing—original draft preparation, M.T., W.M.H. and A.A.; writing—review and editing, A.A., M.T., W.M.H. and A.-A.A.A.E.-S.; visualization, A.A. and M.T.; supervision, A.A.; project administration, A.A.; funding acquisition, A.A, M.T., A.-A.A.A.E.-S. and W.M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Scientific Research Deanship at the Islamic University of Madinah, Saudi Arabia, via project number (RG-1013).
Data Availability Statement
Not applicable.
Conflicts of Interest
There is no conflict of interest to declare. We also have no financial interest to declare.
References
- Secretary-General, U. Progress towards the Sustainable Development Goals: Report of the Secretary-General; UN: New York, NY, USA, 2021. [Google Scholar]
- Abdulrahman, M. Seawater desalination: The strategic choice for Saudi Arabia. Desalination Water Treat. 2012, 51, 1–4. [Google Scholar]
- Ghaffar, A.; Sehgal, S.A.; Fatima, R.; Batool, R.; Aimen, U.; Awan, S.; Batool, S.; Ahmad, F.; Nurulain, S.M. Molecular docking analyses of CYP450 monooxygenases of Tribolium castaneum (Herbst) reveal synergism of quercetin with paraoxon and tetraethyl pyrophosphate: In vivo and in silico studies. Toxicol. Res. 2020, 9, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guidelines for Drinking-Water Quality, 4th Edition, Incorporating the 1st Addendum. 24 April 2017. Available online: https://www.who.int/publications/i/item/9789241549950 (accessed on 24 April 2022).
- Hotta, N. Clinical aspect of chronic arsenic poisoning due to environmental and occupational pollution in and around a small refining spot. JPN J. Const. Med. 1989, 53, 49–70. [Google Scholar]
- Maghraby, M.; Nasr, O.; Hamouda, M. Quality assessment of groundwater at south Al Madinah Al Munawarah area, Saudi Arabia. Environ. Earth Sci. 2013, 70, 1525–1538. [Google Scholar] [CrossRef]
- Ali, I.; Hasan, M.A.; Alharbi, O.M.L. Toxic metal ions contamination in the groundwater, Kingdom of Saudi Arabia. J. Taibah Univ. Sci. 2020, 14, 1571–1579. [Google Scholar] [CrossRef]
- Mohanty, M.; Patra, H.K. Attenuation of Chromium Toxicity by Bioremediation Technology. Rev. Environ. Contam. Toxicol. 2011, 210, 1–34. [Google Scholar]
- Liang, J.; Huang, X.; Yan, J.; Li, Y.; Zhao, Z.; Liu, Y.; Ye, J.; Wei, Y. A review of the formation of Cr(VI) via Cr(III) oxidation in soils and groundwater. Sci. Total Environ. 2021, 774, 145762. [Google Scholar]
- Zhitkovich, A. Chromium in drinking water: Sources, metabolism, and cancer risks. Chem. Res. Toxicol. 2011, 24, 1617–1629. [Google Scholar] [CrossRef]
- Chen, Z.; Song, S.; Wen, Y. Reduction of Cr(VI) into Cr [10] by organelles of Chlorella vulgaris in aqueous solution: An organelle-level attempt. Sci. Total Environ. 2016, 572, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.M.; Viñarta, S.C.; Bernal, A.R.; Cruz, E.L.; Figueroa, L.I. Bioremediation strategies for chromium removal: Current research, scale-up approach and future perspectives. Chemosphere 2018, 208, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, D.; Sukla, L.B.; Sawyer, M.; Rahman, P.K. Recent bioreduction of hexavalent chromium in wastewater treatment: A review. J. Ind. Eng. Chem. 2017, 55, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Daneshvar, E.; Zarrinmehr, M.J.; Kousha, M.; Hashtjin, A.M.; Saratale, G.D.; Maiti, A.; Vithanage, M.; Bhatnagar, A. Hexavalent chromium removal from water by microalgal-based materials: Adsorption, desorption and recovery studies. Bioresour. Technol. 2019, 293, 122064. [Google Scholar] [CrossRef]
- Wołowiec, M.; Komorowska-Kaufman, M.; Pruss, A.; Rzepa, G.; Bajda, T. Removal of Heavy Metals and Metalloids from Water Using Drinking Water Treatment Residuals as Adsorbents: A Review. Minerals 2019, 9, 487. [Google Scholar] [CrossRef] [Green Version]
- Kanmani, P.L.; Aravind, J.; Preston, D. Remediation of chromium contaminants using bacteria. Int. J. Environ. Sci. Technol. 2012, 9, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Spain, O.; Plöhn, M.; Funk, C. The cell wall of green microalgae and its role in heavy metal removal. Physiol. Plant. 2021, 173, 526–535. [Google Scholar] [CrossRef]
- Asha, L.P.; Sandeep, R.S. Review on bioremediation–potential tool for removing environmental pollution. Int. J. Basic Appl. Chem. Sci. 2013, 3, 21–33. [Google Scholar]
- Sharma, J.; Shamim, K.; Dubey, S.K.; Meena, R.M. Metallothionein assisted periplasmic lead sequestration as lead sulfite by Providencia vermicola strain SJ2A. Sci. Total Environ. 2017, 579, 359–365. [Google Scholar] [CrossRef]
- Naik, M.M.; Khanolkar, D.; Dubey, S.K. Lead-resistant Providencia alcalifaciens strain 2EA bioprecipitates Pb+2 as lead phosphate. Lett. Appl. Microbiol. 2013, 56, 99–104. [Google Scholar] [CrossRef]
- Abo-Amer, A.E.; Ramadan, A.B.; Abo-State, M.; Abu-Gharbia, M.A.; Ahmed, H.E. Biosorption of aluminum, cobalt, and copper ions by Providencia rettgeri isolated from wastewater. J. Basic Microbiol. 2013, 53, 477–488. [Google Scholar] [CrossRef]
- Thacker, U.; Parikh, R.; Shouche, Y.; Madamwar, D. Hexavalent chromium reduction by Providencia sp. Process Biochem. 2006, 41, 1332–1337. [Google Scholar] [CrossRef]
- Zimmer, A.L.; Thoden, J.B.; Holden, H.M. Three-dimensional structure of a sugar N-formyltransferase from Francisella tularensis. Protein Sci. 2014, 23, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38, D5–D16. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Kapil, R.; Dhakan, D.B.; Sharma, V.K. MP3: A Software Tool for the Prediction of Pathogenic Proteins in Genomic and Metagenomic Data. PLoS ONE 2014, 9, e93907. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [CrossRef] [Green Version]
- Combet, C.; Jambon, M.; Deléage, G.; Geourjon, C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics 2002, 18, 213–214. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Adiyaman, R.; Maghrabi, A.H.A.; Shuid, A.N.; Brackenridge, D.A.; Nealon, J.O.; Philomina, L.S. IntFOLD: An integrated web resource for high performance protein structure and function prediction. Nucleic Acids Res. 2019, 47, W408–W413. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Pranjaya, P.P.; Thatoi, H. In silico studies on structural, functional, and evolutionary analysis of bacterial chromate reductase family responsible for high chromate bioremediation efficiency. SN Appl. Sci. 2020, 2, 1997. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, Y.; Buchko, G.W.; Varnum, S.M.; Robinson, H.; Squier, T.C.; Long, P.E. Structure determination and functional analysis of a chromate reductase from Gluconacetobacter hansenii. PLoS ONE 2012, 7, e42432. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L., 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2016, 37, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Eswaramoorthy, S.; Poulain, S.; Hienerwadel, R.; Bremond, N.; Sylvester, M.D.; Zhang, Y.B.; Berthomieu, C.; Van Der Lelie, D.; Matin, A. Crystal Structure of ChrR—A Quinone Reductase with the Capacity to Reduce Chromate. PLoS ONE 2012, 7, e36017. [Google Scholar] [CrossRef]
- Cristobal, S.; Zemla, A.; Fischer, D.; Rychlewski, L.; Elofsson, A. A study of quality measures for protein threading models. BMC Bioinform. 2001, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, V.; Klumpler, T.; Marek, J.; Kucera, I. The structural and functional basis of catalysis mediated by NAD(P)H:acceptor Oxidoreductase (FerB) of Paracoccus denitrificans. PLoS ONE 2014, 9, e96262. [Google Scholar] [CrossRef]
- Hanukoglu, I. Proteopedia: Rossmann fold: A beta-alpha-beta fold at dinucleotide binding sites. Biochem. Mol. Biol. Educ. 2015, 43, 206–209. [Google Scholar] [CrossRef]
- Abeln, S.; Feenstra, K.A.; Heringa, J. Protein Three-Dimensional Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Dor, O.; Zhou, Y. Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins Struct. Funct. Bioinform. 2007, 66, 838–845. [Google Scholar] [CrossRef]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, C.; Vullo, A.; Pollastri, G. Protein structural motif prediction in multidimensional phi-psi space leads to improved secondary structure prediction. J. Comput. Biol. 2006, 13, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A. Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Wilding, M.; Hong, N.; Spence, M.; Buckle, A.; Jackson, C.J. Protein engineering: The potential of remote mutations. Biochem. Soc. Trans. 2019, 47, 701–711. [Google Scholar] [CrossRef] [PubMed]
- McCarty, P.L.; Semprini, L. Ground-Water Treatment for Chlorinated Solvents. In Handbook of Bioremediation; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Matin, A. Starvation Promoters of Escherichia coli: Their Function, Regulation, and Use in Bioprocessing and Bioremediation. Ann. N. Y. Acad. Sci. 1994, 721, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; Brugna, M.; Aubert, C.; Bernadac, A.; Bruschi, M. Enzymatic reduction of chromate: Comparative studies using sulfate-reducing bacteria. Key role of polyheme cytochromes c and hydrogenases. Appl. Microbiol. Biotechnol. 2001, 55, 95–100. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Computational framework applied to analyze Cr(VI) to Cr(III) reduction by ChrR from P. alcalifaciens.
Figure 1.
Computational framework applied to analyze Cr(VI) to Cr(III) reduction by ChrR from P. alcalifaciens.

Figure 2.
Analyses of evolution performed by PhyML.

Figure 3.
Multiple sequence alignment of P. alcalifaciens and orthologous protein sequences of ChrR that are substantially comparable. The asterisk at the bottom of the alignment indicates conserved residues; the colon implies conservation between categories of features that are highly comparable; while the period denotes conservation between groups with weakly related features. The highlighted boxes represent identified motifs.
Figure 3.
Multiple sequence alignment of P. alcalifaciens and orthologous protein sequences of ChrR that are substantially comparable. The asterisk at the bottom of the alignment indicates conserved residues; the colon implies conservation between categories of features that are highly comparable; while the period denotes conservation between groups with weakly related features. The highlighted boxes represent identified motifs.

Figure 4.
Jpred and Netsurf P analysis of ChrR (P. alcalifaciens) secondary structure where helices are shown in yellow spirals, sheets in purple arrows, and coils in magenta lines. The letters denote amino acids. The exposed and buried residues are shown in red and blue colored lines, respectively. The disordered regions are determined by the thickness of the gray-colored line.
Figure 4.
Jpred and Netsurf P analysis of ChrR (P. alcalifaciens) secondary structure where helices are shown in yellow spirals, sheets in purple arrows, and coils in magenta lines. The letters denote amino acids. The exposed and buried residues are shown in red and blue colored lines, respectively. The disordered regions are determined by the thickness of the gray-colored line.

Figure 5.
A cartoon illustration of ChrR from P. alcalifaciens was prepared using the BIOVIA Discovery Studio Visualizer, depicting the three motifs in yellow, purple, and red.
Figure 5.
A cartoon illustration of ChrR from P. alcalifaciens was prepared using the BIOVIA Discovery Studio Visualizer, depicting the three motifs in yellow, purple, and red.

Figure 6.
Intra-molecular interactions formed between Cr(VI) and the binding pocket residues of ChrR (P. alcalifaciens). (A) Interaction of Cr(VI) with ChrR wild type, (B) interaction of Cr(VI) with mutant ChrR Glu144Trp, (C) interaction of Cr(VI) with mutant ChrR Gln126Trp, and (D) interaction of Cr(VI) with mutant ChrR Arg82Cys.
Figure 6.
Intra-molecular interactions formed between Cr(VI) and the binding pocket residues of ChrR (P. alcalifaciens). (A) Interaction of Cr(VI) with ChrR wild type, (B) interaction of Cr(VI) with mutant ChrR Glu144Trp, (C) interaction of Cr(VI) with mutant ChrR Gln126Trp, and (D) interaction of Cr(VI) with mutant ChrR Arg82Cys.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physical and chemical characteristics of chromate reductase.
| Parameters | Measures |
|---|---|
| No. of amino acids | 185 |
| Mw | 20,604.39 |
| Theoretical pI | 7.81 |
| Negatively charged residues | 16 |
| Positively charged residues | 17 |
| Ext. coefficient | 30,940 |
| Estimated half-life | 30 h (mammalian reticulocytes, in vitro) |
| IA | 41.23 |
| AI | 82.27 |
| GRAVY | −0.262 |
Note(s): Abbreviations: Mw: molecular weight; IA: instability index; AI: aliphatic index; GRAVY: grand average of hydropathicity.
Table 2.
The predicted virulence factor of chromate reductase from P. alcalifaciens.
| Webserver | Method | Result |
|---|---|---|
| MP3 (Prediction of Pathogenic Proteins in Metagenomic Datasets) | HMM | Non-pathogenic |
| Hybrid | Non-pathogenic | |
| SVM | Non-pathogenic | |
| VirulentPred | Based on Amino acid Composition | 0.8062 (Virulent) |
| Based on Dipeptide Composition | 0.0167 (Virulent) | |
| Based on Higher Order Dipeptide Composition | −0.186 (Non-virulent) | |
| Similarity-Based using PSI-BLAST | 0 No Hits obtained | |
| PSI-BLAST created PSSM Profiles | −0.017 (Non-Virulent) | |
| Cascade of SVMs and PSI-BLAST | 0.6832 (Virulent) | |
| VICMPred | Patterns + Compositions | −1.7423155 |
Table 3.
Evaluation of ChrR from P. alcalifaciens homology-modeled structures.
| Modeling Tool | Residues | ProQ | Global Quality Score | PSVS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG | Max Sub | Verify3D | ProsaII | MolProbity Clashscore | Most Favoured | Additionally Allowed | Generous Allowed | RMSD (Bond Angle) | RMSD (Bond Length) | ||
| Phyre2 | 178 | 8.45 | −0.35 | 0.20 | 0.62 | 88.47 | 92.6 | 4 | 3.4 | 1.4 | 0.011 |
| Geno3D | 174 | 7.96 | −0.35 | 0.19 | 0.49 | 27.86 | 87.1 | 7.6 | 5.3 | 0.9 | 0.004 |
| Robetta | 185 | 8.36 | −0.58 | 0.22 | 0.70 | 2.43 | 89.4 | 9.3 | 1.2 | 1.7 | 0.018 |
| IntFold | 185 | 7.79 | −0.37 | 0.20 | 0.66 | 96.53 | 93.4 | 2.7 | 3.8 | 4.0 | 0.030 |
| I-Tasser | 185 | 8.15 | −0.58 | 0.19 | 0.65 | 8.68 | 86.3 | 11.5 | 2.2 | 2.4 | 0.014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tasleem, M.; Hussein, W.M.; El-Sayed, A.-A.A.A.; Alrehaily, A. Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study. Water 2023, 15, 1142. https://doi.org/10.3390/w15061142
AMA Style
Tasleem M, Hussein WM, El-Sayed A-AAA, Alrehaily A. Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study. Water. 2023; 15(6):1142. https://doi.org/10.3390/w15061142
Chicago/Turabian StyleTasleem, Munazzah, Wesam M. Hussein, Abdel-Aziz A. A. El-Sayed, and Abdulwahed Alrehaily. 2023. "Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study" Water 15, no. 6: 1142. https://doi.org/10.3390/w15061142
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.