
Analysis of Genetic Diversity in Coilia nasus Based on 2b-RAD Simplified Genome Sequencing
by
Yu Li
1,2,
Jianhua Chen
2,
Guangpeng Feng
1,2,3,4,*,
Qingyun Wang
5,
Rulong Xia
5,
Chao Song
1,
Haihua Wang
4 and
Yanping Zhang
4 1
East China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Shanghai 200090, China
2
Jiangsu Key Laboratory of Marine Biotechnology, College of Marine Science and Fisheries, Jiangsu Ocean University, Lianyungang 222005, China
3
Jiangsu Zhongyang Group Co., Ltd., Nantong 226600, China
4
Jiangxi Institute for Fisheries Sciences, Poyang Lake Fisheries Research Centre of Jiangxi Province, Nanchang 330039, China
5
Fisheries Research Institute, Wuhan Academy of Agricultural Sciences, Wuhan 430207, China
*
Author to whom correspondence should be addressed.
Water 2023, 15(6), 1173; https://doi.org/10.3390/w15061173
Submission received: 1 January 2023
/
Revised: 7 March 2023
/
Accepted: 15 March 2023
/
Published: 17 March 2023
(This article belongs to the Special Issue Ecology of Freshwater Fishes)
Abstract
:In the protection of migratory species, Coilia nasus has always been a research topic of very high interest in various countries because of the high economic value and the serious decline of resources. In this study, C. nasus were collected from the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake. By using 2b-RAD simplified genome sequencing technology, 63,110 SNP sites were screened, and the genetic diversity of each population was analyzed with SNP markers. The results showed that (1) the genetic purity of the four groups of populations was not high, with the need to further increase levels of genetic purity; (2) the genetic diversity in the four populations was high, indicating that they had strong adaptability to environmental changes and could easily expand their distribution and resource growth; (3) the FST values (0.112–0.142) of the four populations were higher, indicating that there was moderate genetic differentiation, but no independent population was formed. This study speculated that these migratory species may have the same spawning grounds and pointed out that the gene purity of C. nasus may have been polluted, and it is urgent to improve the purity in the protection of migratory C. nasus.
1. Introduction
Biodiversity refers to all forms of life and their variations in terrestrial, marine and other aquatic ecosystems and their ecological complexes on Earth, including species diversity, genetic diversity and ecosystem diversity [1]. Biodiversity is a necessary prerequisite for the functioning of ecosystems, which is also the material basis for the survival and sustainable development of human society [1]. Affected by human activities, the current global biodiversity is changing at an unprecedented rate. Endemic species are increasingly endangered and extinct, and cosmopolitan species continue to spread and invade. This ecological process causes a decline in the original characteristics of local communities and an increase in regional community similarity, which in turn drives “biological homogenization” [2,3,4,5]. Biological homogenization includes taxonomic, functional and genetic homogenization, and the ecological consequences of biological homogenization are very serious [6,7]. Homogenization of taxonomy will lead to the extinction of endemic indigenous species and change the global species composition; functional homogenization will affect the maintenance of ecosystem functions, thereby reducing the anti-interference ability of biological communities and ecosystems; genetic homogenization will reduce the genetic variation of populations and affect the evolutionary potential of populations and their adaptability to environmental changes [8,9,10]. At present, in order to solve the biodiversity crisis and promote the restoration of biodiversity and resources, especially the diversity and resource restoration of aquatic organisms, countries have introduced many corresponding protection measures to protect aquatic animals such as fish, crabs and shrimp. For example, Australia prohibits commercial fishing in the estuary of New South Wales; China has implemented a ten-year fishing ban in the Yangtze River; Europe encourages selective fishing based on policies [11,12,13]. However, the habitats of migratory species are wide, not only involving rivers and lakes, but also offshore waters [14]. Therefore, single conservation measures of rivers and lakes are not conducive to protecting migratory aquatic animals. C. nasus is a representative migratory fish that is widely distributed in the Yangtze River [15]. In the history of China, C. nasus was an important fisheries resource of the Yangtze River due to the rich population and the relatively stable fishing season [16,17]. However, due to overfishing, water pollution and hydraulic engineering construction, the natural germplasm resource of C. nasus in the Yangtze River has declined sharply in the recent year [17]. The annual yield of C. nasus has been decreasing yearly, and the tendency of individual miniaturization is serious. Notably, the dominant population cannot be formed, so the protection of C. nasus has attracted attention from the world [18,19].
The study of genetic diversity in C. nasus is conducive to protecting the wild population, improving the inheritance of the gene pool specifically for cultured populations, and providing a scientific reference for the restoration of the water ecological environment and stock assessment [20,21]. Genetic diversity is the basis of species diversity and the key evidence for evaluating biological resources. In this regard, many scholars have widely used genetic diversity in the study of species genetic differentiation and achieved remarkable results. For example, a detailed study of Scorpaena spp. in Turkey was carried out using molecular biology, which provided a scientific reference for its resource status and resource restoration. The uniqueness of the Leptopsammia pruvoti population on the Italian coastline was revealed by using 2b-RAD population genomics. The genetic diversity of a Cynoglossus cynoglossus population was analyzed based on 2b-RAD simplified genome sequencing, which pointed out the development direction of artificial reproduction in the future [20,21,22,23,24,25,26,27]. At present, the study of genetic diversity in C. nasus mainly focuses on the population differences in various lakes, aquaculture and the wild, and various smaller water areas of the Yangtze River [22,23,24]. The mitochondrial cytochrome b (Cyt b) gene and control region (D-loop) sequence are used as molecular markers and microsatellite markers to analyze the genetic diversity of C. nasus [24]. Eleven microsatellite loci polymorphisms and mtDNA COI sequences were used to examine the genetic diversity of C. nasus in Japan and China, which showed that the lack of a population genetic structure might result in its amphidromous life cycle, and the geographical distance and habitat fragments might cause isolated populations [25]. Previous studies have found that C. nasus in different lakes have high genetic differentiation; the cultured C. nasus need further breeding and gradually tends to be pure. C. nasus in the Yangtze River is still a species without obvious genetic differentiation [23,24]. However, these studies simply confirm the degree of genetic differentiation among populations of C. nasus, and the study of C. nasus offshore is lacking. 2b-RAD technology is a simplified genome sequencing technology based on type IIB restriction endonuclease [26]. Previous studies have used 2b-RAD technology to analyze the population genetic structure, genetic diversity and genetic relationship between groups of organisms such as Cynoglossus cynoglossus and macaque and showed that it can quickly and comprehensively obtain more markers and has higher typing accuracy, so it can be better applied in the genetic diversity analysis of populations [26,27,28,29,30]. Meanwhile, 2b-RAD technology is more suitable for species sequencing without a reference genome and is one of the most effective and low-cost methods for SNP development in non-model organisms, especially aquatic organisms [29]. Therefore, 2b-RAD sequencing technology can improve the study of the genetic diversity of aquatic biological resources [31].
Genetic differentiation refers to the existence of genetic differences among individuals within a population. This difference might be caused by gene mutations or other factors, so it can measure the genetic diversity of a population, which can also be used to evaluate the evolutionary trend of the population. In this study, 2b-RAD sequencing technology was used for screening the genome for SNP hotspots that can be used as markers of genetic variation and detection of the polymorphism inside of them. Next, a set of SNP markers were used in assessment of genetic differences inside and between C. nasus populations from four different water areas (Poyang Lake: 29°18′ N 116°30′ E, the Taizhou section of the Yangtze River: 32°31′ N 119°77′ E, the Yangtze River Estuary: 31°27′ N 121°95′ E, the Shengsi Sea area: 30°81′ N 122°09′ E). Poyang Lake, located in the middle reaches of the Yangtze River, is the second largest freshwater lake in China and the farthest known spawning ground for C. nasus [14,19]. The Taizhou section of the Yangtze River is located between the Yangtze River Estuary and Poyang Lake. It is the main reproductive migration channel of C. nasus. According to the data, their spawning grounds are also distributed in the shallow water area of the Taizhou River [17]. The estuary of the Yangtze River is an early foraging place for C. nasus. According to previous studies, the brackish waters of the waters and tributaries on Chongming Island (an island in the estuary of the Yangtze River) also disperse their spawning sites [14,19]. The Shengsi Sea area is the confluence of the Yangtze River and the East China Sea, and it has also been proven to be one of the water areas for foraging and fattening of C. nasus [18]. These four waters are very representative. They are all in the Yangtze River system and are connected with each other. However, the relationship between C. nasus living in the four water areas is not clear. The aim of this study is to find the correlation between populations of C. nasus in the four water areas. The results will provide some targeted protection strategies and a scientific reference for global migratory species.
2. Materials and Methods
2.1. Sample Collection and Genomic DNA Extraction
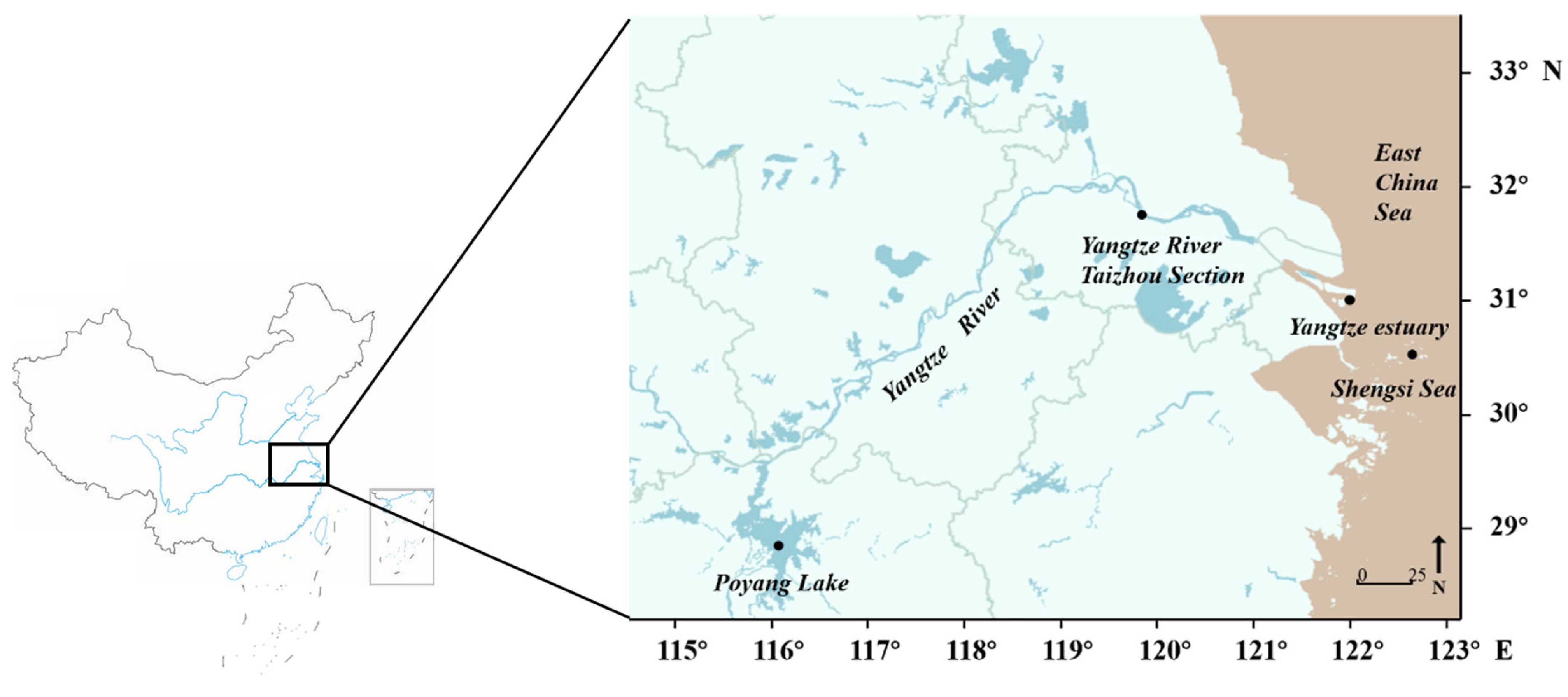
At four sampling sites (the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake), 40 C. nasus individuals were collected using a drift net (Table 1; Figure 1). At each sampling site, individuals with larger maxilla than the head were selected to ensure that samples were river–sea migratory C. nasus by morphological measurement [32]. Moreover, the gender was identified according to the morphological characteristics of gonads, and the stage of gonadal development was determined, and only individuals with stage III gonadal development were collected to reduce sequencing errors [33,34,35,36]. After observing the degree of gonadal reproduction, the C. nasus at stage III of gonadal development was placed in a dry refrigerator and transported back to the laboratory. Subsequently, C. nasus were numbered in the laboratory, and traditional morphological measurements (body weight, body length) were recorded. The small muscle tissue of C. nasus (based on the small muscle below the right dorsal fin of fish) was taken in a 2 mL centrifuge tube and stored at −80 °C. Genomic DNA was successfully extracted from all samples. Two samples from Taizhou could not be sequenced, because they were not well preserved, which were rejected in the subsequent analysis.
RNA-free genomic DNA was extracted from all samples using an animal tissue genomic DNA extraction kit (Sangon Biotech Co., Ltd., Shanghai, China) and eluted with 100 μL of water, as required by the manufacturer. The concentration and quality of extracted DNA were estimated using a NanoDrop UV-Vis spectrophotometer 2000c (Thermo Scientific, Wilmington, DE, USA), and the integrity of DNA was examined using GelRed (Biotium, Fremont, CA, USA; GelRed TM Nucleic AcidStain, 10,000× in Water) on TAE 1 × 1% agarose gel.
2.2. 2b-RAD-Library Preparation and Sequencing Quality Analysis
A tag sequencing library of 40 individuals of C. nasus was constructed using 2b-RAD five-tag tandem technology. Paired-end sequencing was performed on 40 samples on the HiSeq X-Ten platform, and a standard NNN connector was used to construct the library [29,36]. The sequences without BsaXI digestion recognition sites were eliminated, and the low-quality sequences (low-quality sequence definition: the mass fraction of more than 10 bases is less than 20) were eliminated. The sequences with more than 10 consecutive same bases were eliminated, and the original reads were filtered. The average ratio of high-quality reads containing restriction sites in 40 sequencing libraries to the original reads was more than 90%, and the label depth was between 0.02 and 3.21, with an average depth of 0.96, indicating that the sequencing quality of the C. nasus library was good.
2.3. Genome-Wide SNP Screening and Genotyping Analysis
For marker typing with a reference genome, the marker typing is as follows, according to the RAD-typing strategy [37]. Construction of reference sequences: The tags containing BsaXI restriction sites were extracted from the reference genome sequence of C. nasus as reference sequences. The BWA (Burrows–Wheeler Aligner) mem program was used to compare the filtered high-quality data with the reference gene [29,30,31]. SNP genotyping using maximum likelihood was performed [37]. To ensure the accuracy of subsequent data analysis, the classification results were further filtered, and 63,110 classification results were completed.
2.4. Data Processing Method
SNP detection was performed using GATK software (Version 3.7, Broad Institute, Boston, United States) [33]. SNP genotyping results were processed using Genepop software (Version1.0.5). Genetic parameters such as effective number of alleles (Ne), expected heterozygosity (He), observed heterozygosity (Ho), inbreeding coefficient (FIS), nucleotide diversity (Pi) and fixation index (FST) used in assessment of population differentiation were calculated with Vcftools software (Version 0.1.13) [38]. Principal component analysis (PCA) was performed using GCTA software. The phylogenetic tree was constructed by the maximum likelihood algorithm in fastTree software (OSDL: Open Source Development Labs, Inc, Beaverton, OR, USA).
3. Results
3.1. 2b-RAD Sequencing Result Analysis
The results of 2b-RAD sequencing analysis of four populations of C. nasus in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake were as follows: a total of 275,214,800 original reads were obtained, and the average number of sequences per sample was 6,880,370. The high-quality sequences containing BsaXI restriction sites were screened out from each sample, accounting for more than 90% of the original sequences, and the quality of sequencing was good [39].
3.2. Genetic Diversity Analysis of the Four Populations
The genetic diversity of four C. nasus populations in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake was analyzed. In this study, the average expected heterozygosity (He) was 0.146–0.273, and the average observed heterozygosity (Ho) was 0.727–0.854. Moreover, the He was the highest in the YR population (0.273) and the lowest in the TZ population (0.146). The Ho was the highest in the TZ population (0.854) and the lowest in the YR population (0.727). The average nucleotide diversity (Pi) was 0.332, 0.226, 0.219 and 0.177 in YR, SS, PY and TZ, respectively. The average inbreeding coefficient (FIS) was 0.204, 0.271, 0.241 and 0.215 in YR, SS, PY and TZ, respectively (Table 2).
3.3. SNP Mutation Type Analysis
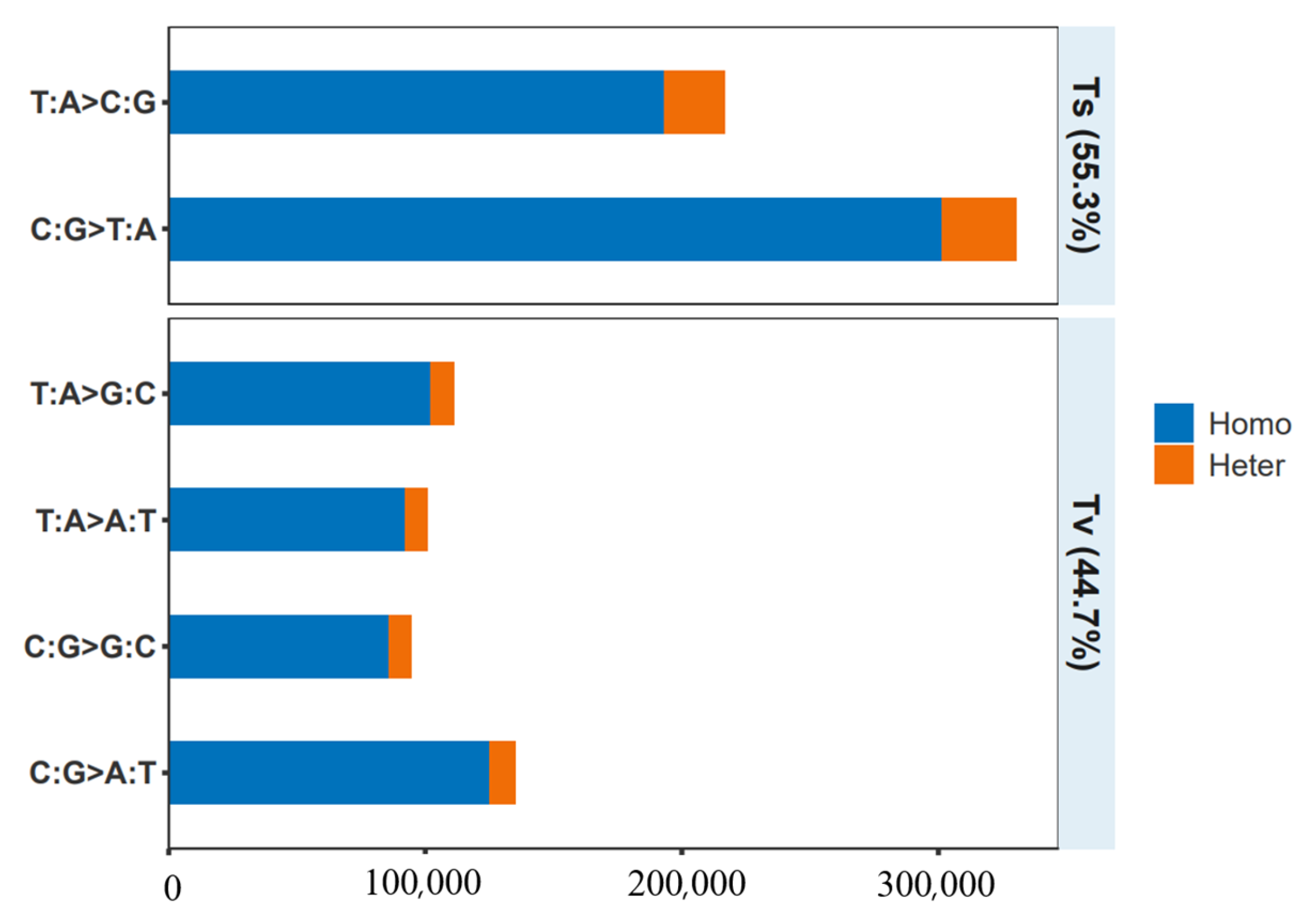
The types of SNP base substitution are divided into two: transition (TS) and transversion (TV). Transition occurs between A and G or C and T, and the transversion occurs between A and C, A and T, G and C, and G and T. The mutation type characteristics of each point in SNP typing results were statistically analyzed. The transformation type is the most numerous, accounting for 55.3% of all base mutation types. The inversion type is the least numerous, accounting for 44.7% of all base mutation types. The ratio of transition to transversion (TS/TV) in SNPs was 1.237 (Figure 2).
3.4. Analysis of Genetic Variation and Differentiation among Populations
The genetic variation and differentiation among four populations of C. nasus in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake were analyzed. In this study, the FST of four C. nasus populations ranged from 0.112 to 0.142, with an average of 0.1235 (Table 3). According to Wright’s division of the genetic differentiation index, the FST value of populations is 0–0.05, and the differentiation is very small, which can be ignored. The FST value of populations was 0.05–0.15, and there was moderate genetic differentiation among the populations. The FST value of the populations was 0.15–0.25, and the genetic differentiation between the populations was large. The populations’ FST value was greater than 0.25, and there was great genetic differentiation among populations [39,40]. Among them, the genetic differentiation coefficient between the Yangtze Estuary and the Shengsi population was 0.112, and the genetic differentiation coefficient between the Poyang Lake and the Taizhou section of the Yangtze River was 0.142.
3.5. Population Genetic Structure Analysis
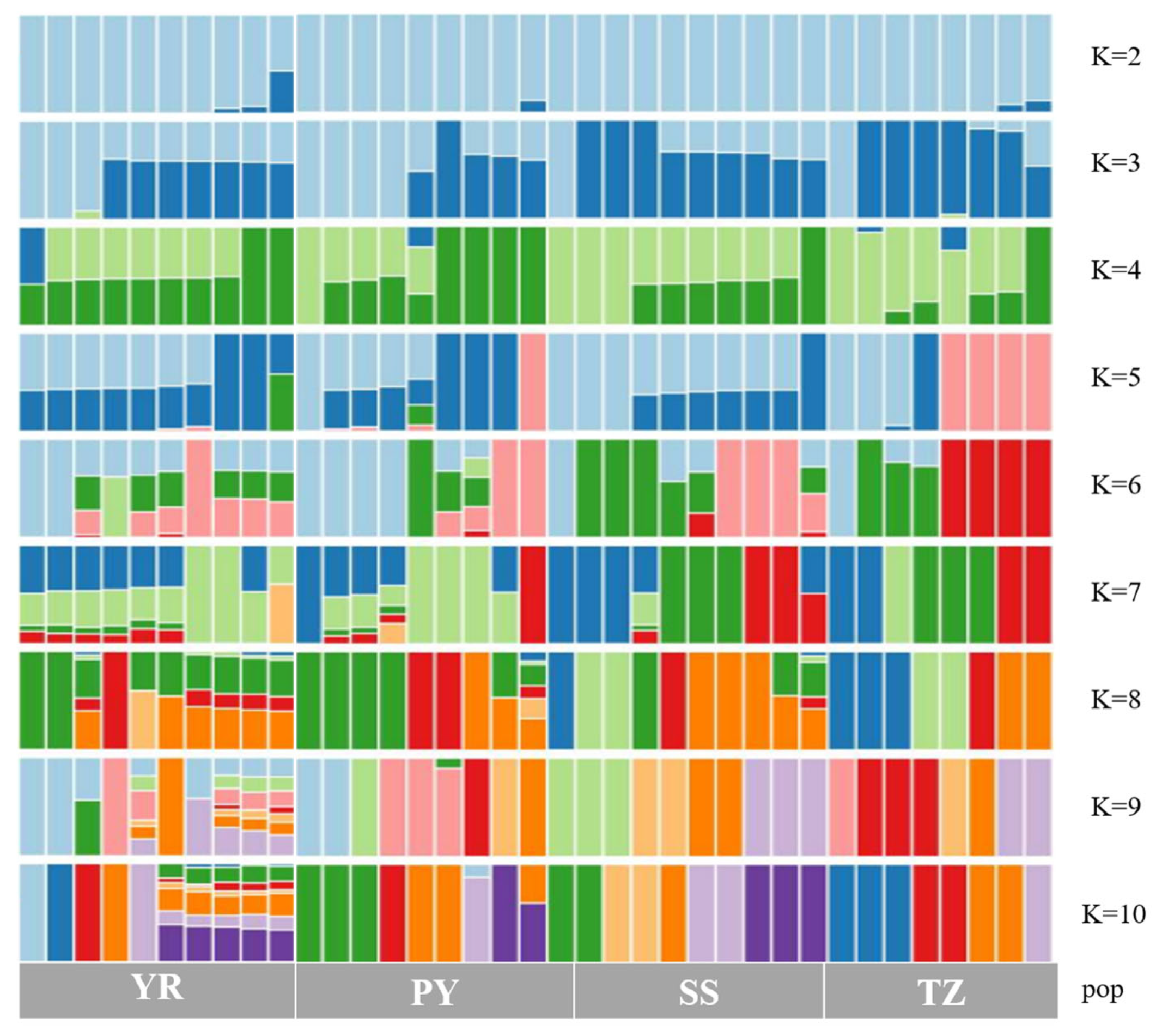
The genetic structure of four C. nasus populations in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake was analyzed. The results showed that the frequency of alleles in the four subgroups fluctuated, indicating that there was a certain degree of genetic differentiation among them (Figure 3).
3.6. Phylogenetic Tree Reconstruction and Principle Component Analysis Based on SNP
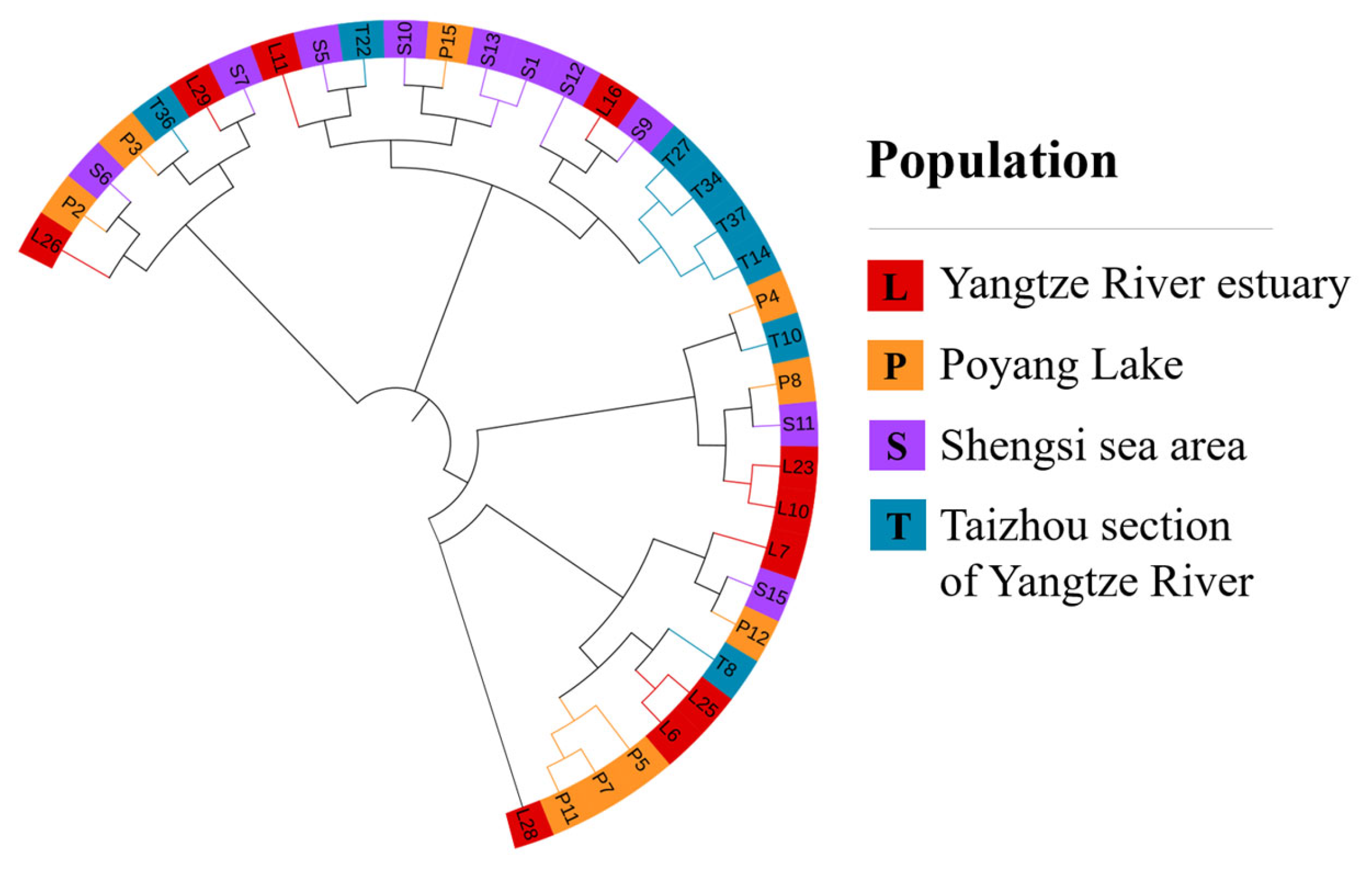
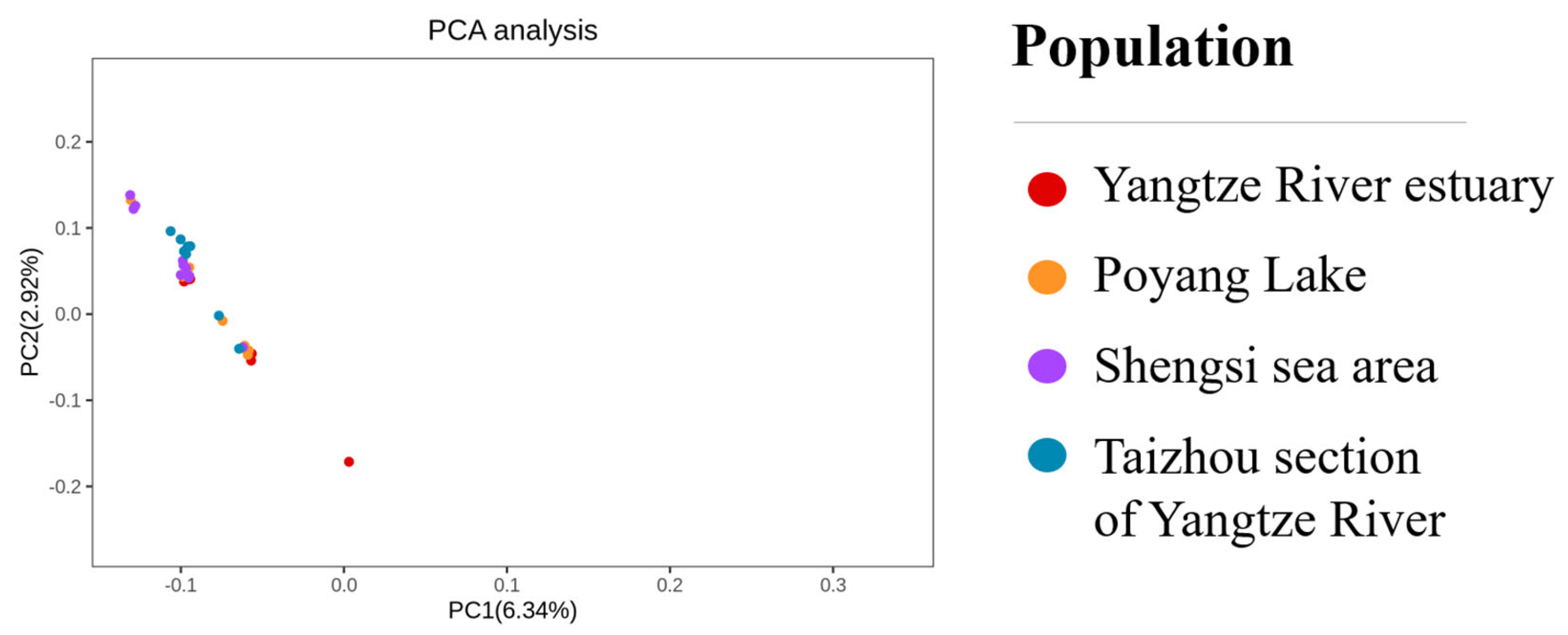
The phylogenetic tree reconstruction and principal component analysis of SNP were carried out in four C. nasus populations in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake. In the phylogenetic tree, except for T14, T27, T34 and T37, other individuals of the four C. nasus populations were hybrid and related (Figure 4). For the principal component scatter plot, only one individual in Poyang Lake was isolated, and the remaining individuals overlap with each other (Figure 5).
4. Discussion
4.1. Genetic Diversity Analysis of Coilia nasus in Four Water Areas
Genetic diversity is the basis of biodiversity and is essential for the survival, reproduction and evolution of species. The genetic diversity of the four C. nasus populations in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River, and Poyang Lake was analyzed in this study. The expected heterozygosity (He), observed heterozygosity (Ho), nucleotide diversity (Pi) and inbreeding coefficient (FIS) reflected the level of genetic diversity in the population. The Ho (0.727–0.854) was much higher than He (0.146–0.273) in four C. nasus populations, indicating that the four C. nasus populations may have been admixtures with other populations, and further purification is needed, especially for the TZ population. Additionally, previous studies have reported the genetic diversity of wild Larimichthys crocea (He: 0.273–0.320, Ho: 0.190–0.253) and wild Apostichopus japonicas (He: 0.318–0.328, Ho: 0.266–0.276) [41,42]. The heterozygosity of the C. nasus population was higher than that of L. crocea and A. japonicas, so it is speculated that the genetic diversity of the four C. nasus populations was at a high level and had a high potential for genetic variation. The FIS of four C. nasus populations (0.204–0.271) were all positive, suggesting that the degree of inbreeding was severe, especially in the SS population. Previous studies have shown that there are freshwater settled C. nasus populations in the Yangtze River, and these individual resources are much larger than migratory individuals; these settled populations may interbreed with migratory populations [43,44]. Therefore, while restoring migratory individual resources, it is also necessary to protect the purity of C. nasus. The following measures are recommended to be implemented: reducing the proliferation and release activities to avoid gene exchange between river–sea migratory individuals and artificially cultured individuals and protecting the currently known spawning grounds to avoid the use of unified spawning grounds for freshwater individuals and river–sea migratory individuals.
4.2. SNP Mutation Type Analysis
The resulting ratio of transitions and transversions (TS/TV) in this study is 1.237, which is much larger than the theoretical probability ratio of TS/TV of 0.5, which is called “transition bias” [45,46,47]. The results of 2b-RAD sequencing in this study showed that there was a significant conversion bias in SNP polymorphism types. According to previous studies, there are great differences in the ratio of conversion and transversion between different species. The ratio of transition to transversion of 2b-RAD in Fenneropenaeus chinensis was 1.402 [48]. The ratio of conversion to transversion in transcriptome sequencing of Pinctada fucata was 0.5 [49]. The ratio of transition to transversion in the sequencing results of abalone was 2.33 [50]. In summary, the conversion bias was due to the different habitats of the populations and evolution and different selection pressures, which led to this phenomenon [48,49,50,51]. It is worth noting that previous studies have shown that there are multiple spawning grounds for swimming crabs in the Yangtze River, which begin to migrate and reproduce at different times [39,52,53]. Therefore, the reason for the TS/TV being less than 0.5 was speculated to be that the four C. nasus populations in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake have different times and migration routes during reproductive migration, resulting in different environmental pressures experienced in evolution.
4.3. Genetic Differentiation of Coilia nasus in Four Water Areas
The genetic differentiation index (FST) is an important parameter to evaluate the genetic differentiation in the populations. The results of population genetic structure analysis showed that there were different gene frequencies in the four populations of the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake, indicating that there was a certain degree of genetic differentiation. Phylogenetic tree and principal component analysis were constructed based on SNP. The results of the phylogenetic tree showed that except for T14, T27, T34 and T37, which were clustered into a small branch, other individuals were interspersed and clustered into one group. The results of principal component analysis showed that the four C. nasus populations overlapped with each other, except for one individual in the Yangtze River estuary. In summary, this study shows that there is a moderate degree of genetic differentiation between each sample, and there is a certain range of gene exchange among each other. Previous studies have shown that the water areas suitable for the spawning of C. nasus in the Yangtze River are scarce [39]. Moreover, there is still a large number of freshwater sedentary individuals in the Yangtze River, and their breeding seasons and freshwater habitats are similar to those of migratory individuals [39]. Therefore, it was speculated that there were two reasons for a certain range of gene exchange among the four populations of C. nasus in the Shengsi Sea area, the Yangtze River estuary, the Taizhou section of the Yangtze River and Poyang Lake: (1) Some of the spawning grounds of these four groups are the same freshwater areas; (2) Freshwater settled individuals or other migratory individuals acted as a medium between them, resulting in a certain range of gene exchange.
4.4. Restoration of Coilia nasus Resources
This study found that the genetic diversity of C. nasus in the Yangtze River waters is high, and it has great environmental adaptability and evolutionary potential. It has high germplasm resources protection value and development and utilization potential. It was also found that there is a Yangtze River C. nasus gene pollution phenomenon. Therefore, in future resource recovery, stocking juveniles in hatchery conditions and release should be reduced, and the genetic distinctness of C. nasus populations needs to be protected by natural recovery. To rely on natural recovery, it is necessary to increase the protection of water areas. C. nasus belongs to river–sea migratory fish, which is distributed not only in fresh water but also in offshore water. Therefore, in the future, we should further restore and protect the resources of C. nasus in the offshore water on the basis of the ten-year fishing ban plan of the Yangtze River, so as to better promote the recovery and sustainable utilization of their resources and provide a scientific reference for the global migratory species.
5. Conclusions
2b-RAD technology with high accuracy was used to analyze the genetic diversity of C. nasus in Poyang Lake, the Taizhou section of the Yangtze River, the Yangtze Estuary and the Shengsi Sea area. Firstly, it is confirmed that the 2b-RAD method can be applied to the C. nasus species, which provides a new method for subsequent C. nasus research. Secondly, the results emphasized that there were abundant genetic variation and significant genetic structure among C. nasus populations, which provides a scientific basis for future resource recovery. Finally, it was speculated that the four C. nasus populations might reproduce in the same spawning ground, although there were temporal and spatial differences in migratory breeding. Some recommendations were made for the restoration of C. nasus resources, protecting the purity of species and expanding the scope of protected water areas. From the perspective of future management of river–sea migratory species, the proposal is effective in promoting resource recovery of river–sea migratory species, and it can be better applied to restore global river–sea migratory species resources. Moreover, the use of stable isotopes in fish otoliths to explore their spawning grounds has been reported. Therefore, stable isotope studies can be carried out on the otoliths of juvenile C. nasus in the future, and the results of this study can be reasonably supplemented.
Author Contributions
Conceptualization, Y.L., G.F., Q.W. and R.X.; Data curation, Y.L., H.W. and G.F.; Formal analysis, Y.L. and G.F.; Funding acquisition, G.F.; Project administration, G.F.; Visualization, Y.L. and Y.Z.; Writing—original draft, Y.L., J.C. and C.S.; Writing—review and editing, G.F. and Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Key R & D Program Key Special Project (2019YFD0901205); Jiangsu Innovation and Entrepreneurship Team Project (JSSCTD202120); Species Resources Protection Project of the Ministry of Agriculture and Rural Affairs.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article. Further information is available upon request from the corresponding author.
Acknowledgments
We would like to express our great appreciation to the editor and the reviewers for their constructive comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pimm, S.L.; Russell, G.J.; Gittleman, J.L.; Brooks, T.M. The future of biodiversity. Science 1995, 269, 347–350. [Google Scholar] [CrossRef] [Green Version]
- McKinney, M.L.; Lockwood, J.L. Biotic homogenization: A few winners replacing many losers in the next mass extinction. Trends Ecol. Evol. 1999, 14, 450–453. [Google Scholar] [CrossRef]
- Olden, J.D. Biotic homogenization: A new research agenda for conservation biogeography. J. Biogeogr. 2006, 33, 2027–2039. [Google Scholar] [CrossRef]
- Magurran, A.E.; Dornelas, M.; Moyes, F.; Gotelli, N.J.; McGill, B. Rapid biotic homogenization of marine fish assemblages. Nat. Commun. 2014, 6, 8405. [Google Scholar] [CrossRef] [Green Version]
- Iacarella, J.C.; Adamczyk, E.; Bowen, D.; Chalifour, L.; Eger, A.; Heath, W.; Helms, S.; Hessing-Lewis, M.; Hunt, B.P.V.; MacInnis, A.; et al. Anthropogenic disturbance homogenizes seagrass fish communities. Glob. Chang. Biol. 2018, 24, 1904–1918. [Google Scholar] [CrossRef]
- Dar, P.A.; Reshi, Z.A. Components, processes and consequences of biotic homogenization: A review. Contemp. Probl. Ecol. 2014, 7, 123–136. [Google Scholar] [CrossRef]
- Rosenblad, K.C.; Sax, D.F. A new framework for investigating biotic homogenization and exploring future trajectories: Oceanic island plant and bird assemblages as a case study. Ecography 2017, 40, 1040–1049. [Google Scholar] [CrossRef]
- Storfer, A. Gene flow and endangered species translocations: A topic revisited. Biol. Conserv. 1999, 87, 173–180. [Google Scholar] [CrossRef]
- Beisner, B.E.; Ives, A.R.; Carpenter, S.R. The effects of an exotic fish invasion on the prey communities of two lakes. J. Anim. Ecol. 2003, 72, 331–342. [Google Scholar] [CrossRef]
- Petsch, D.K. Causes and consequences of biotic homogenization in freshwater ecosystems. Int. Rev. Hydrobiol. 2016, 101, 113–122. [Google Scholar] [CrossRef]
- Salim, M.; William, G. Ban on commercial fishing in the estuarine waters of New South Wales, Australia: Community consultation and social impacts. Environ. Impact Assess. Rev. 2007, 28, 214–225. [Google Scholar]
- Ruilong, W.; Yi, H.; Fei, F. Need to shift in river-lake connection scheme under the “ten-year fishing ban” in the Yangtze River, China. Ecol. Indic. 2022, 143, 109434. [Google Scholar]
- Condie, H.; Grant, A.; Catchpole, T. Incentivising selective fishing under a policy to ban discards; lessons from European and global fisheries. Mar. Policy 2014, 45, 287–292. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, H.; Shen, X.; Shimasaki, Y.; Ohshima, Y.; Yang, J. Life History variations among different populations of Coilia nasus along the Chinese Coast inferred from otolith microchemistry. J. Fac. Agric. Kyushu Univ. 2014, 59, 383–389. [Google Scholar] [CrossRef]
- Yuan, C.M. Spawning migration of Coilia nasus. Bull. Biol. 1987, 12, 1–3. [Google Scholar]
- Yuan, C.M.; Qin, A.; Liu, R.; Lin, J. Discussion on the subspecies classification of Coilia in the middle and lower reaches of the Yangtze River and southeast coastal provinces. J. Nanjing Univ. 1980, 3, 67–82. [Google Scholar]
- Duan, J.; Zhang, H.; Liu, K.; Xu, D.; Zhang, M.; Shi, W. An overview of Coilia ectenes in Jiangsu section of the Yangtze River. Agric. Sci. Technol. 2012, 13, 1950–1954. [Google Scholar]
- Li, Y.; Chen, J.; Feng, G.; Yang, J.; Zhao, F.; Shen, C.; Song, C.; Jiang, T. Otolith microchemistry assessment: Evidence of migratory Coilia nasus of Yangtze River living in the Shengsi Sea area. Fishes 2022, 7, 172. [Google Scholar] [CrossRef]
- Jiang, T.; Yang, J.; Xuan, Z.Y.; Chen, X.; Liu, H. Preliminary report on the effects of resource recovery on anadromous Coilia nasus in Poyang Lake under the National 10-Year Fishing Ban. Prog. Fish. Sci. 2022, 43, 24–30. [Google Scholar]
- Yedier, S.; Bostancı, D. Molecular and otolith shape analyses of Scorpaena spp. in the Turkish seas. Turk. J. Zool. 2022, 46, 78–92. [Google Scholar]
- Pei, L.M.; Gong, Y.Y.; Huang, B.Y.; Zhang, Q.; Xu, S. Genetic diversity analysis of long-horned silver bass in northern South China Sea based on mitochondrial COI. Mar. Fish. 2020, 42, 257–265. [Google Scholar]
- Zhang, J.; Gao, S.; Shi, Y.; Yan, Y.; Liu, Q. Full-length transcriptome of anadromous Coilia nasus using single molecule real-time (SMRT) sequencing. Aquac. Fish. 2022, 7, 420–426. [Google Scholar] [CrossRef]
- Chen, X.; Song, P.; Xia, J.; Guo, J.; Shi, Y.; Zhong, Y.; Li, M. Evolutionarily conserved boule and dazl identify germ cells of Coilia nasus. Aquac. Fish. 2023, 8, 244–251. [Google Scholar] [CrossRef]
- Wei, G.; Xu, G.; Gu, R.; Li, J.; Xu, P. Studies on the genetic diversity of farmed and wild populations of Coilia nasus by analysing mitochondrial DNA Cyt b genes. Acta Agric. Univ. Jiangxiensis 2012, 34, 1216–1221+1244. [Google Scholar]
- Yang, J.-Q.; Hsu, K.-C.; Zhou, X.-D.; Kuo, P.-H.; Lin, H.-D.; Liu, D.; Bao, B.-L.; Tang, W.-Q. New insights on geographical/ecological populations within Coilia nasus (Clupeiformes: Engraulidae) based on mitochondrial DNA and microsatellites. Mitochondrial DNA Part A 2018, 29, 158–164. [Google Scholar] [CrossRef]
- Min, F.; Xu, F.; Huang, S.; Wu, R.; Zhang, L.; Wang, J. Genetic diversity of Chinese laboratory macaques based on 2b-RAD simplified genome sequencing. J. Med. Primatol. 2022, 51, 101–107. [Google Scholar] [CrossRef]
- Hu, J.; Ren, H.Y. RAD sequencing technology and its application in aquatic biology. Aquat. Sci. 2018, 37, 125–132. [Google Scholar]
- Boscari, E.; Abbiati, M.; Badalamenti, F.; Bavestrello, G.; Benedetti-Cecchi, L.; Cannas, R.; Cau, A.; Cerrano, C.; Chimienti, G.; Costantini, F.; et al. A population genomics insight by 2b-RAD reveals population’ uniqueness along the Italian coastline in Leptopsammia pruvoti (Scleractinia, Dendrophylliidae). Biodivers. Res. 2019, 25, 1101–1117. [Google Scholar]
- Jia, L.; Zhang, B.; Liu, K.; Zheng, D.; Wang, X. Analysis of Population Genetic Diversity of Cynoglossus cynoglossus based on 2b-RAD simplified genome sequencing. Open J. Fish. Res. 2017, 4, 125–133. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van Inghelandt, D.; Melchinger, A.E.; Lebreton, C.; Stich, B. Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor. Appl. Genet. 2010, 120, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xie, S.; Li, Z.; Gong, W.; He, W. Gonad development of an anadromous fish Coilia ectenes (Engraulidae) in lower reach of Yangtze River, China. Fish. Sci. 2007, 73, 1224–1230. [Google Scholar]
- Zhu, P.; He, L.; Li, Y.; Huang, W.; Xi, F.; Lin, L.; Zhi, Q.; Zhang, W.; Tang, Y.T.; Geng, C.; et al. OTG-snpcaller: An Optimized Pipeline Based on TMAP and GATK for SNP Calling from Ion Torrent Data. PLoS ONE 2014, 9, e97507. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Wan, J.; Gu, R.; Zhang, C.; Xu, P. Morphological and histological studies on ovary development of Coilia nasus under artificial farming conditions. J. Fish. Sci. China 2011, 18, 537–546. [Google Scholar] [CrossRef]
- Xu, G.C.; Wan, J.J.; Gu, R.B.; Zhang, C.; Xu, P. Histological studies on testis development of Coilia nasus under artificial farming conditions. J. Huazhong Agric. Univ. 2012, 31, 247–252. [Google Scholar]
- Wen, H.B. Development of gonads in Coilia nasus from the Yangtze River and artificial pond. Chin. J. Zool. 2009, 44, 111–117. [Google Scholar]
- Fu, X.; Dou, J.; Mao, J.; Su, H.; Jiao, W.; Zhang, L.; Hu, X.; Huang, X.; Wang, S.; Bao, Z. RADtyping: An Integrated Package for Accurate De Novo Codominant and Dominant RAD Genotyping in Mapping Populations. PLoS ONE 2013, 8, e79960. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Duan, J.R.; Zhou, Y.F.; Xu, D.P.; Zhang, M.Y.; Liu, K.; Shi, Y.; Wei, Q.W.; Fang, D.A. Ovary transcriptome profiling of Coilia nasus during spawning migration stages by Illumina sequencing. Mar. Genom. 2015, 21, 17–19. [Google Scholar] [CrossRef]
- Curnow, R.N.; Wright, S. Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, Y.; Zhang, J.; Zhu, A.; Wu, C.; Liu, L.; Lin, Z.; Dong, Y. Population structure of large yellow croaker (Larimichthys crocea) revealed by single nucleotide polymorphisms. Biochem. Syst. Ecol. 2015, 63, 136–142. [Google Scholar] [CrossRef]
- Dong, Y.; Li, Q.; Zhong, X.; Kong, L. Development of gene-derived SNP markers and their application for the assessment of genetic diversity in wild and cultured populations in sea cucumber, Apostichopus japonicus. J. World Aquac. Soc. 2016, 47, 873–888. [Google Scholar] [CrossRef]
- Xu, Z.Q.; Ge, J.C.; Huang, C.; Dou, H.X.; Pan, J.L.; Xia, A.J. Taxonomy of shortjaw tapertail anchovy Coilia brachygnathus by jaw length and mitochondrial Cytochrome b gene analysis. J. Dalian Fish. Univ. 2009, 24, 242–246. [Google Scholar]
- Wang, S.; Wang, B.; Hu, M.; Wang, F.; Wu, Z. The complete mitochondrial genome of Coilia brachygnathus (Clupeiformes: Engraulidae: Coilinae). Mitochondrial DNA Part A 2016, 27, 4084–4085. [Google Scholar] [CrossRef]
- Collins, D.W.; Jukes, T.H. Rates of transition and transversion in coding sequences since the human-rodent divergence. Genomics 1994, 20, 386–396. [Google Scholar] [CrossRef]
- Gojobori, T.; Li, W.-H.; Graur, D. Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol. 1982, 18, 360–369. [Google Scholar] [CrossRef]
- Li, W.-H.; Wu, C.-I.; Luo, C.-C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984, 21, 58–71. [Google Scholar] [CrossRef]
- Wang, F.; Meng, X.H.; Fu, Q.; Luan, S.; Sui, J. Analysis of genetic diversity in three generations of breeding population of Fenneropenaeus chinensis based on reduced-representation genome sequencing. Prog. Fish. Sci. 2020, 41, 68–76. [Google Scholar]
- Wang, Z.L.; Ding, Y.; Xu, Y.H.; Wang, B.; Zhang, J.D. SNP discovery and functional annotation in transcriptome datasets from hemocytes of Pinctada fucata. Oceanol. Limnol. Sin. 2018, 49, 403–412. [Google Scholar]
- Wang, L.X.; Ke, C.H.; Wang, Z.Y.; Liu, B.; Cai, M.Y.; Wang, Y.L. Sequence analysis of mitochondrial 16S rRNA gene and molecular phylogeny of eight species of abalones in genus Haliotis. J. Fish. Sci. China. 2006, 13, 167–173. [Google Scholar]
- Verity, R.; Nichols, R.A. What is genetic differentiation, and how should we measure it-GST, D, neither or both? Mol. Ecol. 2014, 23, 4216–4225. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, H.; Lu, M.; Chen, T.; Yang, J. A possible connectivity among estuarine tapertail anchovy (Coilia nasus) populations in the Yangtze River, Yellow Sea, and Poyang Lake. Estuaries Coasts 2016, 39, 1762–1768. [Google Scholar] [CrossRef]
- Liu, D.; Li, Y.; Tang, W.; Yang, J.; Guo, H.; Zhu, G.; Li, H. Population structure of Coilia nasus in the Yangtze River revealed by insertion of short interspersed elements. Biochem. Syst. Ecol. 2014, 54, 103–112. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Sampling site map of Coilia nasus.

Figure 2.
SNP mutation spectrum analysis.

Figure 3.
Population structure of Coilia nasus by SNPS Analysis (K = 2–10). Note: Each column in the picture represents an individual, and the length of different color fragments represents the proportion of an ancestor in the individual’s genome.
Figure 3.
Population structure of Coilia nasus by SNPS Analysis (K = 2–10). Note: Each column in the picture represents an individual, and the length of different color fragments represents the proportion of an ancestor in the individual’s genome.

Figure 4.
The phylogenetic tree.

Figure 5.
Principal component scatter plot.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Basic information of each population sample of Coilia nasus.
| Locations | Acronym | Sampling Time | Number of Samples | Body length (mm) | Body Weight (g) | Geographical Coordinates | Sex (Female/Male) |
|---|---|---|---|---|---|---|---|
| Shengsi Sea area | SS | April 2021 | 10 | 246.36 | 30.17 | 30°81′ N 122°09′ E | 5/5 |
| 251.16 | 41.22 | ||||||
| 262.49 | 32.32 | ||||||
| 278.53 | 57.56 | ||||||
| 265.28 | 41.24 | ||||||
| 339.82 | 144.49 | ||||||
| 334.72 | 106.9 | ||||||
| 282.54 | 63.6 | ||||||
| 339.82 | 144.49 | ||||||
| 345.64 | 157.03 | ||||||
| Yangtze River estuary | YR | April 2021 | 10 | 157.31 | 10.39 | 31°27′ N 121°95′ E | 5/5 |
| 234.08 | 30.11 | ||||||
| 250.24 | 33.33 | ||||||
| 248.03 | 37.16 | ||||||
| 254.92 | 33.74 | ||||||
| 243.76 | 28.79 | ||||||
| 265.50 | 48.57 | ||||||
| 267.17 | 43.32 | ||||||
| 251.49 | 37.09 | ||||||
| 355.29 | 137.87 | ||||||
| Taizhou section of Yangtze River | TZ | May 2021 | 10 | 218.99 | 25.13 | 32°31′ N 119°77′ E | 5/5 |
| 277.62 | 53.71 | ||||||
| 234.39 | 34.1 | ||||||
| 275.90 | 66.50 | ||||||
| 283.55 | 64.9 | ||||||
| 304.74 | 84.5 | ||||||
| 306.93 | 83.5 | ||||||
| 303.86 | 77.3 | ||||||
| 311.33 | 71.2 | ||||||
| 356.27 | 133 | ||||||
| Poyang Lake | PY | July 2021 | 10 | 213.94 | 16.38 | 29°18′ N 116°30′ E | 5/5 |
| 280.34 | 48.4 | ||||||
| 242.42 | 31.2 | ||||||
| 231.68 | 28.6 | ||||||
| 238.11 | 29.8 | ||||||
| 245.01 | 35.2 | ||||||
| 271.02 | 36.2 | ||||||
| 233.83 | 28.42 | ||||||
| 268.93 | 53.33 | ||||||
| 286.45 | 78.62 |
Table 2.
Analysis of genetic polymorphism parameters of SNP loci in Coilia nasus populations.
| Population | He | Ho | Pi | FIS |
|---|---|---|---|---|
| YR | 0.273 | 0.727 | 0.332 | 0.204 |
| SS | 0.203 | 0.780 | 0.226 | 0.271 |
| PY | 0.195 | 0.805 | 0.219 | 0.241 |
| TZ | 0.146 | 0.854 | 0.177 | 0.215 |
Table 3.
Genetic differentiation coefficient of four populations of Coilia nasus.
| Population | YR | SS | PY | TZ |
|---|---|---|---|---|
| YR | 0.112 | 0.115 | 0.129 | |
| SS | 0.113 | 0.131 | ||
| PY | 0.142 | |||
| TZ |
Note: The FST value of the populations is 0–0.05; the differentiation is very small and can be ignored. The FST value of the populations was 0.05–0.15, and there was moderate genetic differentiation among the populations. The FST value of the populations was 0.15–0.25, and the genetic differentiation between the populations was large. The populations’ FST value was greater than 0.25, and there was great genetic differentiation among populations.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Y.; Chen, J.; Feng, G.; Wang, Q.; Xia, R.; Song, C.; Wang, H.; Zhang, Y. Analysis of Genetic Diversity in Coilia nasus Based on 2b-RAD Simplified Genome Sequencing. Water 2023, 15, 1173. https://doi.org/10.3390/w15061173
AMA Style
Li Y, Chen J, Feng G, Wang Q, Xia R, Song C, Wang H, Zhang Y. Analysis of Genetic Diversity in Coilia nasus Based on 2b-RAD Simplified Genome Sequencing. Water. 2023; 15(6):1173. https://doi.org/10.3390/w15061173
Chicago/Turabian StyleLi, Yu, Jianhua Chen, Guangpeng Feng, Qingyun Wang, Rulong Xia, Chao Song, Haihua Wang, and Yanping Zhang. 2023. "Analysis of Genetic Diversity in Coilia nasus Based on 2b-RAD Simplified Genome Sequencing" Water 15, no. 6: 1173. https://doi.org/10.3390/w15061173
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.